---
title: "Database Schema"
subtitle: "Architecture + ERDs for the v8 Marine Atlas DuckDB databases (serve · sdm · merge · spp)"
---
The **v8 Marine Atlas** pipeline flows through four DuckDB databases. The one consumers query is
the tiny **`serve.duckdb`** — a set of *views* over partitioned Parquet on S3 (KB on disk, never a
multi-GB monolith). It is fed by the multi-GB build database **`sdm.duckdb`**, which in turn is
assembled from the merge intermediate **`merge.duckdb`** and the taxonomy authority **`spp.duckdb`**.
```{r}
#| label: setup
#| include: false
librarian::shelf(
DBI, duckdb, DT, dplyr, fs, glue, purrr, tibble,
quiet = T)
source(here::here("libs/paths.R"))
# v8 database locations (read-only) ----
dir_atlas <- glue("{dir_big_v}/marine-atlas")
db_paths <- list(
serve = glue("{dir_atlas}/serve/serve.duckdb"), # PUBLIC: views over S3 parquet
sdm = sdm_db, # build DB (multi-GB)
merge = glue("{dir_atlas}/merge.duckdb"), # two-surface merge intermediate
spp = spp_db) # taxonomy authority (WoRMS/GBIF/ITIS/IUCN/BOTW)
open_db <- function(p) if (file_exists(p)) dbConnect(duckdb(dbdir = p, read_only = TRUE)) else NULL
con_serve <- open_db(db_paths$serve)
con_sdm <- open_db(db_paths$sdm)
con_merge <- open_db(db_paths$merge)
con_spp <- open_db(db_paths$spp)
# core (published) tables per DB — the auto-generated "Tables" sections document these only, so
# build-only scratch/experimental tables (hex*, wtmp*, v7_*, mc_* staging) don't clutter the doc.
core_tables <- list(
serve = c("cell","model","model_cell","taxon","dataset","metric","cell_metric",
"zone","zone_cell","zone_metric","native_asset"),
sdm = c("cell","model","model_cell","taxon","dataset","metric","cell_metric",
"zone","zone_cell","zone_metric","native_asset"),
merge = c("taxon","taxon_model","taxon_flags","taxon_cell","taxon_cell_global","taxon_er",
"listing","model_cell","mc_parts","mkey_map","turtle_src","us_cells"),
spp = c("worms","botw","gbif","itis","iucn_redlist",
"worms_vernacular","botw_vernacular","gbif_vernacular","itis_vernacular","iucn_vernacular"))
# expected schema (update when columns change; check_schema flags drift vs the live DB) ----
serve_expected <- list(
cell = c("cell_id","lon","lat","depth_mean","depth_min","depth_max","oxy_b_mean","oxy_mean",
"prim_prod_mean","ice_con_ann","salinity_b_mean","salinity_mean","sbt_an_mean",
"sst_an_mean","fao_area_m","area_km2","in_usa","in_pra"),
model = c("mdl_key","mdl_id","ds_key","sp_id","sci_name","common_name","er_score","sp_cat"),
model_cell = c("mdl_key","mdl_id","cell_id","val","value"),
taxon = c("taxon_authority","taxon_id","ms_merge_key","scientific_name","iucn_code","n_models",
"n_datasets","worms_is_marine","worms_is_extinct","extrisk_code","er_score","is_mmpa",
"is_mbta","is_bcc","common_name","n_cells","n_ocean","n_usa","n_pra","range_km2",
"range_usa_km2","n_global","is_valid_global","is_valid_usa","is_valid_pra","pct_marine",
"us_endemism","rarity","sp_cat","is_marine","pct_marine_bl","in_v7","is_er_spatial"),
dataset = c("ds_key","name_short","response_type","source_broad","temporal_res","native_format",
"sort_order","qmd","name_original","description","citation","source_detail","regions",
"taxa_groups","year_pub","date_obs_beg","date_obs_end","date_env_beg","date_env_end",
"link_info","link_download","link_metadata","links_other","spatial_res_deg",
"date_created","name_display","value_info","is_mask","global_mask_priority"),
metric = c("metric_seq","metric_key","description"),
cell_metric = c("cell_id","metric_seq","val","value"),
zone = c("zone_seq","tbl","fld","val","value"),
zone_cell = c("zone_seq","cell_id","pct_covered"),
zone_metric = c("zone_seq","metric_seq","val","value"),
native_asset = c("ms_merge_key","mdl_key","ds_key","asset_type","representation","asset_url",
"rescale_min","rescale_max","colormap","source_layer","xmin","xmax","ymin","ymax"))
# sdm shares the same core-table columns as serve (serve is a view over sdm's Parquet export) except
# model_cell/cell_metric/zone*, where serve adds the `value` alias for titiler; reuse the same list.
sdm_expected <- serve_expected
sdm_expected$model_cell <- c("mdl_key","cell_id","val")
sdm_expected$cell_metric <- c("cell_id","metric_seq","val")
sdm_expected$zone <- c("zone_seq","tbl","fld","val")
sdm_expected$zone_metric <- c("zone_seq","metric_seq","val")
merge_expected <- list(
taxon = serve_expected$taxon[!serve_expected$taxon %in% c("in_v7","is_er_spatial")], # v7 flags added at score_zones
taxon_model = c("mdl_key","taxon_authority","taxon_id","ms_merge_key"),
taxon_flags = c("ms_merge_key","has_am","has_range"),
taxon_cell = c("mdl_key","n_cells","n_ocean","n_usa","n_pra","range_km2","range_usa_km2"),
taxon_cell_global = c("mdl_key","n_global"),
taxon_er = c("ms_merge_key","extrisk_code","er_score","is_mmpa","is_mbta","is_bcc"),
listing = c("sci","nmfs_esa","is_mmpa","fws_esa","is_bcc","is_mbta"),
model_cell = c("mdl_key","cell_id","value"),
mc_parts = c("mkey_id","ms_merge_key","ds_key","cell_id","val"),
mkey_map = c("ms_merge_key","mkey_id"),
turtle_src = c("ms_merge_key","ds_key","cell_id","val"),
us_cells = c("cell_id"))
spp_expected <- list(
worms = c("taxonID","scientificNameID","acceptedNameUsageID","parentNameUsageID","scientificName",
"acceptedNameUsage","parentNameUsage","kingdom","phylum","class","order","family","genus",
"subgenus","specificEpithet","infraspecificEpithet","taxonRank","scientificNameAuthorship",
"nomenclaturalCode","taxonomicStatus","nomenclaturalStatus","isMarine","isFreshwater",
"isTerrestrial","isExtinct","isBrackish"),
worms_vernacular = c("taxonID","vernacularName","source","language","isPreferredName"),
botw = c("taxonID","scientificName","redlist_code","kingdom","phylum","class","order","family",
"family_common_name","subfamily","tribe","taxonomic_authority","taxonomic_sources",
"acceptedNameUsage","acceptedNameUsageID"),
botw_vernacular = c("taxonID","vernacularName","isPreferredName"),
gbif = c("taxonID","datasetID","parentNameUsageID","acceptedNameUsageID","originalNameUsageID",
"scientificName","scientificNameAuthorship","canonicalName","genericName",
"specificEpithet","infraspecificEpithet","taxonRank","nameAccordingTo","namePublishedIn",
"taxonomicStatus","nomenclaturalStatus","taxonRemarks","kingdom","phylum","class",
"order","family","genus"),
gbif_vernacular = c("taxonID","vernacularName","language","country","countryCode","sex",
"lifeStage","source"),
itis = c("taxonID","parentNameUsageID","acceptedNameUsageID","scientificName",
"scientificNameAuthorship","taxonRank","taxonomicStatus","nomenclaturalStatus",
"completeness","namePublishedIn","namePublishedInYear"),
itis_vernacular = c("taxonID","vernacularName","language"),
iucn_redlist = c("taxon_scientific_name","sis_taxon_id","year_published","latest",
"possibly_extinct","possibly_extinct_in_the_wild","url","red_list_category_code",
"assessment_id","code","code_type","scopes_description","scopes_code",
"red_list_category_code_gom","year_published_gom","assessment_id_gom"),
iucn_vernacular = c("taxonID","isPreferredName","language","vernacularName"))
# helper: flag schema drift vs expected (only over the core tables) ----
check_schema <- function(con, expected, core, db_label) {
if (is.null(con)) { cat(glue("::: {{.callout-note}}\n`{db_label}` not found locally — skipped.\n:::\n\n")); return(invisible()) }
actual_cols <- dbGetQuery(con, "
SELECT table_name, column_name FROM information_schema.columns
WHERE table_schema='main' ORDER BY table_name, ordinal_position")
actual <- split(actual_cols$column_name, actual_cols$table_name)
actual <- actual[intersect(names(actual), core)] # only compare documented core tables
msgs <- character()
new_tables <- setdiff(intersect(names(actual), core), names(expected))
missing_tables <- setdiff(intersect(names(expected), core), names(actual))
if (length(new_tables)) msgs <- c(msgs, glue("**Undocumented core tables:** {paste(new_tables, collapse=', ')}"))
if (length(missing_tables)) msgs <- c(msgs, glue("**Missing tables:** {paste(missing_tables, collapse=', ')}"))
for (tbl in intersect(names(actual), names(expected))) {
new_cols <- setdiff(actual[[tbl]], expected[[tbl]])
missing_cols <- setdiff(expected[[tbl]], actual[[tbl]])
if (length(new_cols)) msgs <- c(msgs, glue("**`{tbl}`** new columns: {paste(new_cols, collapse=', ')}"))
if (length(missing_cols)) msgs <- c(msgs, glue("**`{tbl}`** missing columns: {paste(missing_cols, collapse=', ')}"))
}
if (length(msgs)) {
cat("::: {.callout-warning}\n")
cat(glue("## {db_label} schema drift — update the ERD + expected list in schema.qmd\n\n"))
cat(paste("-", msgs, collapse = "\n"), "\n:::\n\n")
} else cat(glue("::: {{.callout-tip appearance='simple'}}\n`{db_label}` matches the documented schema.\n:::\n\n"))
}
# helper: infer column description ----
infer_desc <- function(col) {
known <- c(
cell_id="global 0.05° grid cell id (1:ncell)", mdl_key="stable model id {ds_key}|{sp_id}",
mdl_id="dense integer model id (serving partition key; renumbers per release)",
ms_merge_key="merged-taxon model id ms_merge|{authority}:{id}", ds_key="dataset key",
metric_seq="metric sequence id", zone_seq="zone sequence id", sp_id="species id within dataset",
sp_key="species key in source dataset", sp_cat="taxonomy-based scoring category",
taxon_id="taxon id within its authority", taxon_authority="taxonomic authority (worms/botw/…)",
sci_name="scientific name", scientific_name="scientific name", common_name="common name",
val="value (suitability 0-100 / er_score on range cells)", value="titiler alias of val",
er_score="extinction-risk score (0-100, most-protective)", extrisk_code="extinction-risk code",
iucn_code="IUCN Red List category", redlist_code="IUCN Red List category",
is_mmpa="MMPA-protected (all WoRMS Mammalia)", is_mbta="MBTA-protected (FWS §10.13 birds)",
is_bcc="Bird of Conservation Concern", is_marine="passes the marine-relevance cull",
is_valid_global="valid on the WHOLE global range (drives the app's all-species list)",
is_valid_usa="has ≥1 merged cell in US waters (n_usa>0)", is_valid_pra="present in a Program Area",
in_usa="cell in US study area", in_pra="cell in a BOEM Program Area", in_v7="in v7's scored set",
is_er_spatial="ER baked into cell val (turtles) so not re-scored", n_cells="merged cells (any)",
n_ocean="merged ocean cells", n_usa="merged US cells", n_pra="merged Program-Area cells",
n_global="whole-range merged cells (→ is_valid_global)", range_km2="merged range area (km²)",
range_usa_km2="US merged range area (km²)", pct_marine="% of range that is ocean",
pct_marine_bl="% marine from the BirdLife range", us_endemism="% of ocean range in US waters",
rarity="rarity class from range_km2", n_models="# raw models merged", n_datasets="# datasets merged",
has_am="taxon has an AquaMaps model", has_range="taxon has a range polygon (globally)",
representation="native (original) | model (0.05° gridded)", asset_type="COG | PMTiles",
asset_url="public S3/titiler URL", rescale_min="colormap min", rescale_max="colormap max",
colormap="titiler colormap name", pct_covered="% of cell covered by the zone",
area_km2="cell area (km²)", depth_mean="mean depth (m)", sst_an_mean="annual mean SST",
mkey_id="dense merge-key id", worms_is_marine="marine per WoRMS", worms_is_extinct="extinct per WoRMS",
native_format="native SDM format", global_mask_priority="mask precedence", is_mask="mask layer",
isMarine="marine (WoRMS)", isExtinct="extinct (WoRMS)", scientificName="scientific name",
vernacularName="common name", taxonRank="taxonomic rank", taxonomicStatus="taxonomic status")
if (col %in% names(known)) return(known[[col]])
if (grepl("_id$|ID$", col)) return("identifier")
if (grepl("_key$", col)) return("key")
if (grepl("_seq$", col)) return("sequence id")
if (grepl("^is_|^in_|^has_|^is[A-Z]", col)) return("boolean flag")
if (grepl("^n_", col)) return("count")
if (grepl("_mean$", col)) return("mean value")
if (grepl("_km2$", col)) return("area (km²)")
if (grepl("^date_", col)) return("date")
if (grepl("_code$", col)) return("code")
if (grepl("^link", col)) return("URL")
if (grepl("_name", col)) return("name")
""
}
# helper: table info as a DataTable (introspects the live DB) ----
tbl_info <- function(con, tbl_name) {
cols <- dbGetQuery(con, glue("
SELECT column_name, data_type FROM information_schema.columns
WHERE table_schema='main' AND table_name='{tbl_name}' ORDER BY ordinal_position"))
# COUNT can be slow/unavailable for views over S3 (no httpfs/creds) — never let it halt the render
n_rows <- tryCatch(dbGetQuery(con, glue('SELECT COUNT(*) n FROM "{tbl_name}"'))$n, error = function(e) NA_real_)
big <- is.na(n_rows) || n_rows > 5e7 # skip per-column scans on huge/uncounted tables
stats <- map_dfr(seq_len(nrow(cols)), function(i) {
col <- cols$column_name[i]; dtype <- cols$data_type[i]
col_q <- paste0('"', col, '"'); is_float <- grepl("DOUBLE|FLOAT|DECIMAL|NUMERIC", dtype, ignore.case = TRUE)
range_str <- NA_character_; top5_str <- NA_character_
if (!big) {
if (grepl("INT|DOUBLE|FLOAT|DECIMAL|NUMERIC|DATE|TIMESTAMP", dtype, ignore.case = TRUE)) {
mn <- if (is_float) glue("ROUND(MIN({col_q}),4)") else glue("MIN({col_q})")
mx <- if (is_float) glue("ROUND(MAX({col_q}),4)") else glue("MAX({col_q})")
rng <- tryCatch(dbGetQuery(con, glue('SELECT {mn}::VARCHAR mn, {mx}::VARCHAR mx FROM "{tbl_name}"')), error = function(e) NULL)
if (!is.null(rng) && !is.na(rng$mn)) range_str <- glue("[{rng$mn}, {rng$mx}]")
}
samp <- if (n_rows > 5e6) "USING SAMPLE 1000000" else ""
vex <- if (is_float) glue("ROUND({col_q},4)") else col_q
top5 <- tryCatch(dbGetQuery(con, glue('SELECT {vex}::VARCHAR val, COUNT(*) n FROM "{tbl_name}" {samp} GROUP BY {vex} ORDER BY n DESC LIMIT 5')), error = function(e) NULL)
if (!is.null(top5) && nrow(top5)) {
top5$val <- ifelse(nchar(top5$val) > 40, paste0(substr(top5$val, 1, 37), "..."), top5$val)
top5_str <- paste(glue("{top5$val} ({format(top5$n, big.mark=',')})"), collapse = "; ")
}
}
tibble(column = col, type = dtype, description = infer_desc(col), range = range_str, top_values = top5_str)
})
cap <- if (is.na(n_rows)) glue("{tbl_name} — view (row count via S3, not fetched)")
else glue("{tbl_name} — {format(n_rows, big.mark=',')} rows{if (big) ' (large: per-column stats skipped)' else ''}")
datatable(stats, caption = cap,
options = list(scrollX = TRUE, pageLength = 40, dom = "ft"), rownames = FALSE, escape = FALSE)
}
# helper: render the "Tables" panel-tabset for a DB's core tables ----
tables_panel <- function(con, tbls, prefix) {
present <- intersect(tbls, dbListTables(con))
src <- map_chr(present, function(tn) knitr::knit_child(text = c(
glue("### {tn}\n"), "```{r}", glue('#| label: {prefix}-tbl-{tn}'), "#| echo: false",
glue('tbl_info(con_{prefix}, "{tn}")'), "```", ""), envir = environment(), quiet = TRUE))
cat("::: {.panel-tabset}\n\n"); cat(src, sep = "\n"); cat("\n:::\n")
}
```
## Architecture
```{mermaid}
%%| label: fig-architecture
%%| fig-cap: "v8 Marine Atlas data flow across the four DuckDB databases (click to zoom)"
flowchart TB
src["ingest_*.qmd — one per source<br/>AquaMaps · BirdLife · IUCN/FWS/NMFS ranges · SWOT turtles"]
spp[("<b>spp.duckdb</b><br/>WoRMS · GBIF · ITIS<br/>IUCN · BirdLife<br/><i>taxonomy authority</i>")]
merge[("<b>merge.duckdb</b><br/>taxon · taxon_model · taxon_flags<br/>mc_parts · model_cell<br/><i>two-surface merge intermediate</i>")]
sdm[("<b>sdm.duckdb</b> — build (multi-GB)<br/>cell · model · model_cell · taxon<br/>metric · cell_metric · zone* · native_asset")]
s3[("<b>S3</b> marine-atlas/v8/<br/>tables/ · serve/ · dist_merged/ · registry/")]
serve[("<b>serve.duckdb</b> — KB<br/><i>VIEWs over S3 Parquet</i>")]
titiler["titiler-v8<br/>tile server"]
stac["STAC catalog"]
apps["species · scores<br/>Shiny apps"]
src -->|"dist/dataset={ds_key}/*.parquet<br/>(mdl_key, cell_id, val)"| merge
spp -.->|"sp_cat · is_marine · WoRMS ids"| merge
merge -->|"merge_taxon → score_zones →<br/>score_cell_metrics → score_zone_metrics"| sdm
sdm -->|"release_marine-atlas"| s3
s3 --> serve
serve --> titiler
s3 --> stac
titiler --> apps
stac --> apps
classDef db fill:#1e3a5f,stroke:#3bc9db,color:#e6edf3;
class spp,merge,sdm,serve db;
```
**Key conventions.** The public identifier is the stable **`mdl_key`** (`{ds_key}|{sp_id}`, pipe
separator; merged taxa are `ms_merge|{authority}:{id}`). The dense integer **`mdl_id`** is a serving
optimization only (partition-prunes titiler reads) and renumbers per release, so it never appears in
URLs. Values live in a **`val`** column (`value` is a DuckDB reserved word); serving views expose a
`value` alias for titiler. Scoring runs over `in_usa` cells; `is_valid_global` uses the whole-range
cell tally so species outside the US still list in the app.
---
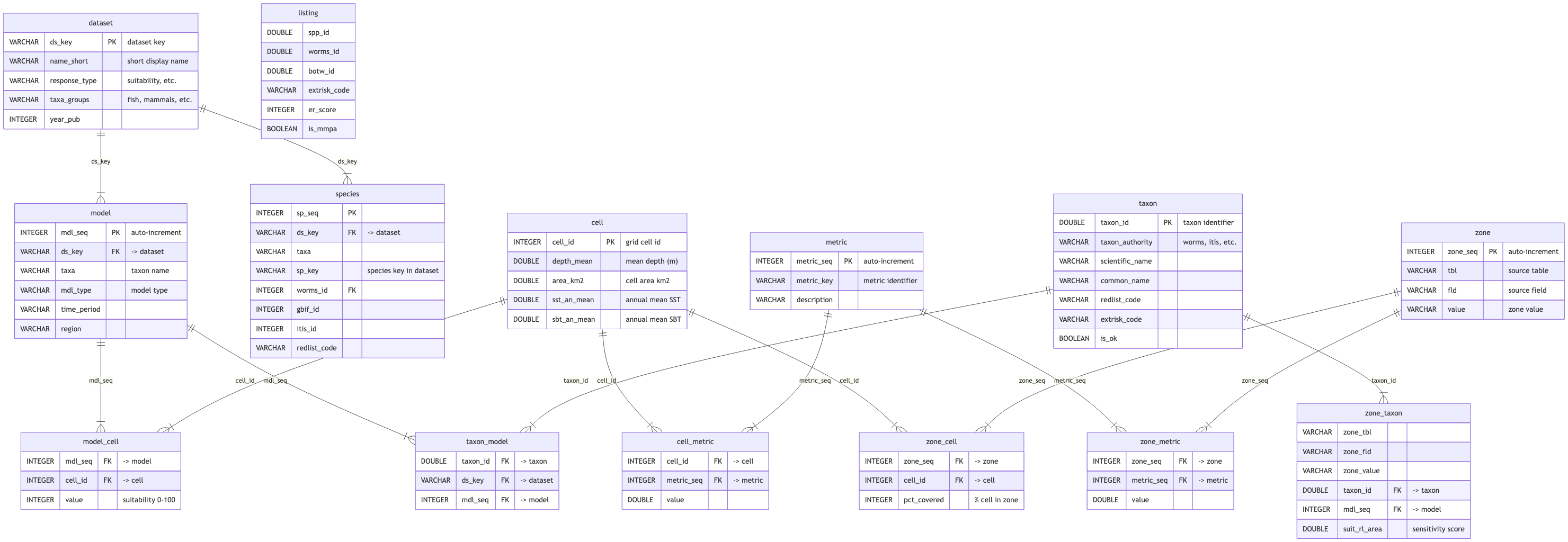
# serve.duckdb — public serving surface
The database consumers query: a few KB of **views** over the partitioned Parquet published to
`s3://oceanmetrics.io-public/marine-atlas/v8/`. titiler-v8 resolves `mdl_key → mdl_id` and reads a
single `serve/model_cell/mdl_id=*/…parquet` partition per tile (anonymous HTTP range read).
```{r}
#| label: serve-changes
#| echo: false
#| output: asis
check_schema(con_serve, serve_expected, core_tables$serve, "serve.duckdb")
```
## Schema
```{mermaid}
%%| label: fig-serve-erd
%%| fig-cap: "serve.duckdb entity relationship diagram (click to zoom)"
%%| fig-width: 12
erDiagram
dataset {
VARCHAR ds_key PK "dataset key"
VARCHAR name_short "short display name"
VARCHAR response_type "suitability, range, …"
VARCHAR native_format "native SDM format"
}
model {
VARCHAR mdl_key PK "stable {ds_key}|{sp_id}"
INTEGER mdl_id "dense serving id"
VARCHAR ds_key FK "-> dataset"
VARCHAR sci_name
DOUBLE er_score
VARCHAR sp_cat
}
model_cell {
VARCHAR mdl_key FK "-> model"
INTEGER mdl_id "serving partition key"
INTEGER cell_id FK "-> cell"
DOUBLE val "suitability 0-100"
}
cell {
INTEGER cell_id PK "0.05° grid id"
DOUBLE lon
DOUBLE lat
DOUBLE area_km2
BOOLEAN in_usa
BOOLEAN in_pra
}
taxon {
VARCHAR ms_merge_key PK "merged model id"
VARCHAR taxon_authority
VARCHAR taxon_id
VARCHAR scientific_name
DOUBLE er_score
VARCHAR sp_cat
BOOLEAN is_valid_global "whole-range validity"
BOOLEAN is_valid_usa "n_usa>0"
}
native_asset {
VARCHAR ms_merge_key FK "-> taxon"
VARCHAR mdl_key FK "-> model"
VARCHAR ds_key FK "-> dataset"
VARCHAR asset_type "COG | PMTiles"
VARCHAR representation "native | model"
VARCHAR asset_url "public S3/titiler URL"
}
metric {
INTEGER metric_seq PK
VARCHAR metric_key
}
cell_metric {
INTEGER cell_id FK "-> cell"
INTEGER metric_seq FK "-> metric"
DOUBLE val
}
zone {
INTEGER zone_seq PK
VARCHAR tbl "program areas / ecoregions"
VARCHAR val "zone key"
}
zone_cell {
INTEGER zone_seq FK "-> zone"
INTEGER cell_id FK "-> cell"
INTEGER pct_covered
}
zone_metric {
INTEGER zone_seq FK "-> zone"
INTEGER metric_seq FK "-> metric"
DOUBLE val
}
dataset ||--|{ model : ds_key
model ||--|{ model_cell : mdl_key
model ||--o{ native_asset : mdl_key
taxon ||--o{ native_asset : ms_merge_key
cell ||--|{ model_cell : cell_id
cell ||--|{ cell_metric : cell_id
cell ||--|{ zone_cell : cell_id
metric ||--|{ cell_metric : metric_seq
metric ||--|{ zone_metric : metric_seq
zone ||--|{ zone_cell : zone_seq
zone ||--|{ zone_metric : zone_seq
```
## Tables
Every table above is a **view** (`SELECT * FROM read_parquet('s3://…/tables/{table}.parquet')`), so
its columns mirror the `sdm.duckdb` core tables documented next (with live row counts + value ranges)
— plus, on the serving views, the titiler `value` alias of `val` and the `mdl_id` join on
`model_cell`. Row counts here would require an anonymous S3 scan and are omitted.
---
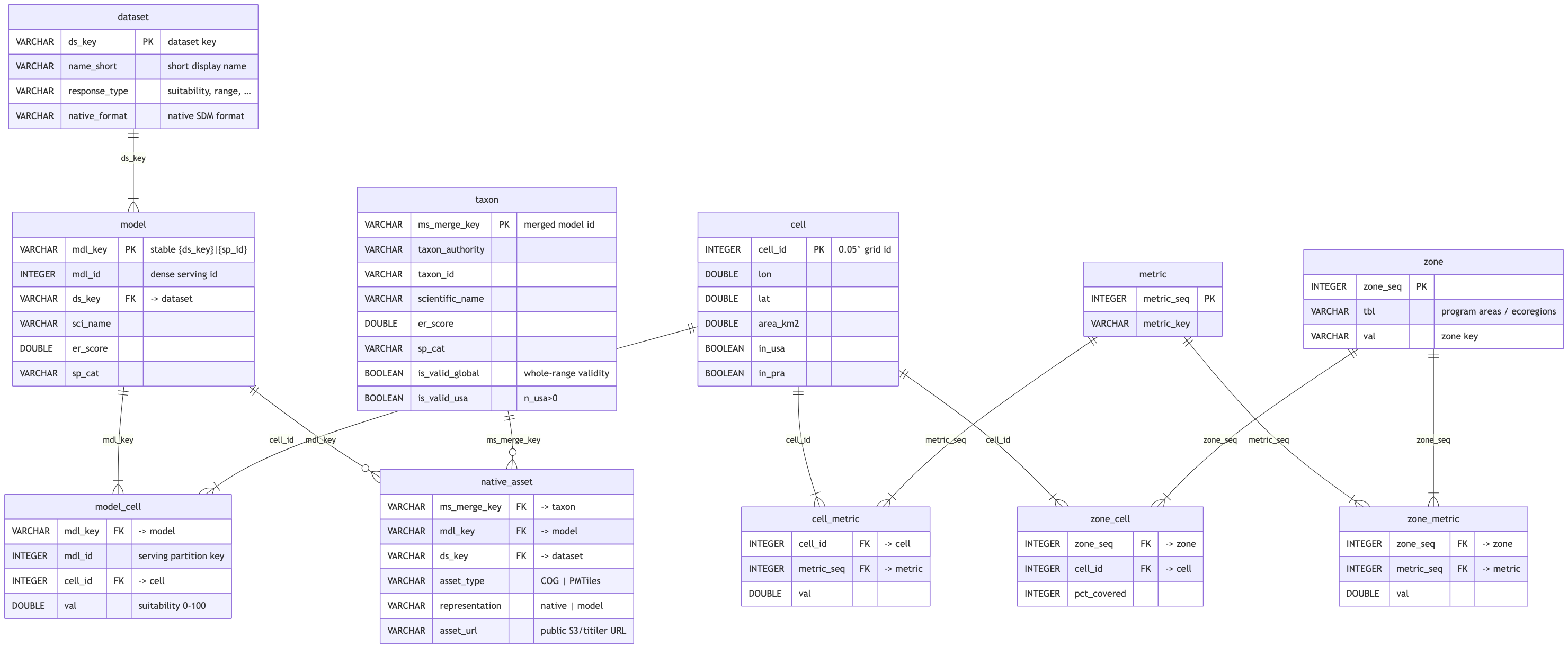
# sdm.duckdb — build database
The multi-GB database `release_marine-atlas` exports to S3. Same core tables as `serve` (serve is a
view over its Parquet); `model_cell` here is `(mdl_key, cell_id, val)` — the `mdl_id` join and
`value` alias are added at serve time. Also holds build-only scratch/experimental tables
(`hex*`, `wtmp*`, `v7_cat`, `v7_ok`) not shown here.
```{r}
#| label: sdm-changes
#| echo: false
#| output: asis
check_schema(con_sdm, sdm_expected, core_tables$sdm, "sdm.duckdb")
```
## Tables
```{r}
#| label: sdm-tables
#| output: asis
#| echo: false
if (!is.null(con_sdm)) tables_panel(con_sdm, core_tables$sdm, "sdm") else cat("_sdm.duckdb not found locally._")
```
---
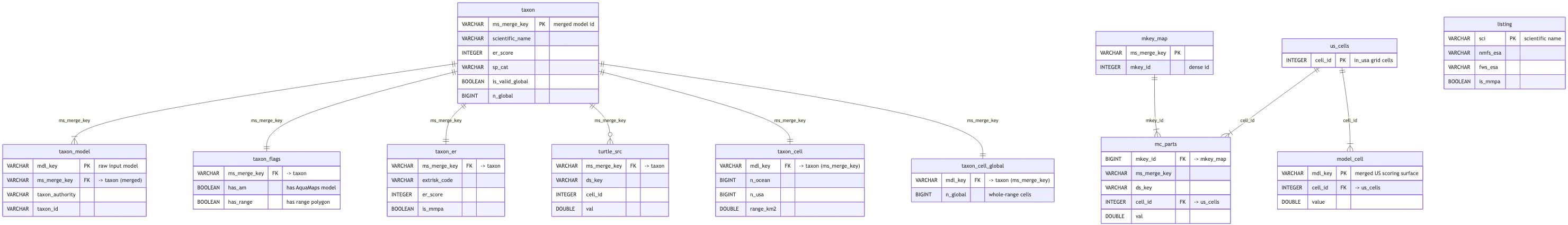
# merge.duckdb — two-surface merge intermediate
`merge_models` writes both surfaces here: a **global** viz surface (`am ∪ range`) and the
**US-scoped** v7-faithful scoring surface (`model_cell`). `merge_taxon` derives per-taxon validity
(`taxon`, from `taxon_cell` + `taxon_cell_global`), governing extinction risk (`taxon_er`), and the
`has_am`/`has_range` flags (`taxon_flags`) that drive the no-EEZ AquaMaps constraint. `score_zones`
copies `taxon` from here into `sdm.duckdb`.
```{r}
#| label: merge-changes
#| echo: false
#| output: asis
check_schema(con_merge, merge_expected, core_tables$merge, "merge.duckdb")
```
## Schema
```{mermaid}
%%| label: fig-merge-erd
%%| fig-cap: "merge.duckdb entity relationship diagram (click to zoom)"
%%| fig-width: 11
erDiagram
taxon_model {
VARCHAR mdl_key PK "raw input model"
VARCHAR ms_merge_key FK "-> taxon (merged)"
VARCHAR taxon_authority
VARCHAR taxon_id
}
taxon {
VARCHAR ms_merge_key PK "merged model id"
VARCHAR scientific_name
INTEGER er_score
VARCHAR sp_cat
BOOLEAN is_valid_global
BIGINT n_global
}
taxon_flags {
VARCHAR ms_merge_key FK "-> taxon"
BOOLEAN has_am "has AquaMaps model"
BOOLEAN has_range "has range polygon"
}
taxon_cell {
VARCHAR mdl_key FK "-> taxon (ms_merge_key)"
BIGINT n_ocean
BIGINT n_usa
DOUBLE range_km2
}
taxon_cell_global {
VARCHAR mdl_key FK "-> taxon (ms_merge_key)"
BIGINT n_global "whole-range cells"
}
taxon_er {
VARCHAR ms_merge_key FK "-> taxon"
VARCHAR extrisk_code
INTEGER er_score
BOOLEAN is_mmpa
}
mc_parts {
BIGINT mkey_id FK "-> mkey_map"
VARCHAR ms_merge_key
VARCHAR ds_key
INTEGER cell_id FK "-> us_cells"
DOUBLE val
}
model_cell {
VARCHAR mdl_key PK "merged US scoring surface"
INTEGER cell_id FK "-> us_cells"
DOUBLE value
}
mkey_map {
VARCHAR ms_merge_key PK
INTEGER mkey_id "dense id"
}
turtle_src {
VARCHAR ms_merge_key FK "-> taxon"
VARCHAR ds_key
INTEGER cell_id
DOUBLE val
}
us_cells {
INTEGER cell_id PK "in_usa grid cells"
}
listing {
VARCHAR sci PK "scientific name"
VARCHAR nmfs_esa
VARCHAR fws_esa
BOOLEAN is_mmpa
}
taxon ||--|{ taxon_model : ms_merge_key
taxon ||--|| taxon_flags : ms_merge_key
taxon ||--|| taxon_er : ms_merge_key
taxon ||--o{ turtle_src : ms_merge_key
taxon ||--|| taxon_cell : ms_merge_key
taxon ||--|| taxon_cell_global : ms_merge_key
mkey_map ||--|{ mc_parts : mkey_id
us_cells ||--|{ mc_parts : cell_id
us_cells ||--|{ model_cell : cell_id
```
## Tables
```{r}
#| label: merge-tables
#| output: asis
#| echo: false
if (!is.null(con_merge)) tables_panel(con_merge, core_tables$merge, "merge") else cat("_merge.duckdb not found locally._")
```
---
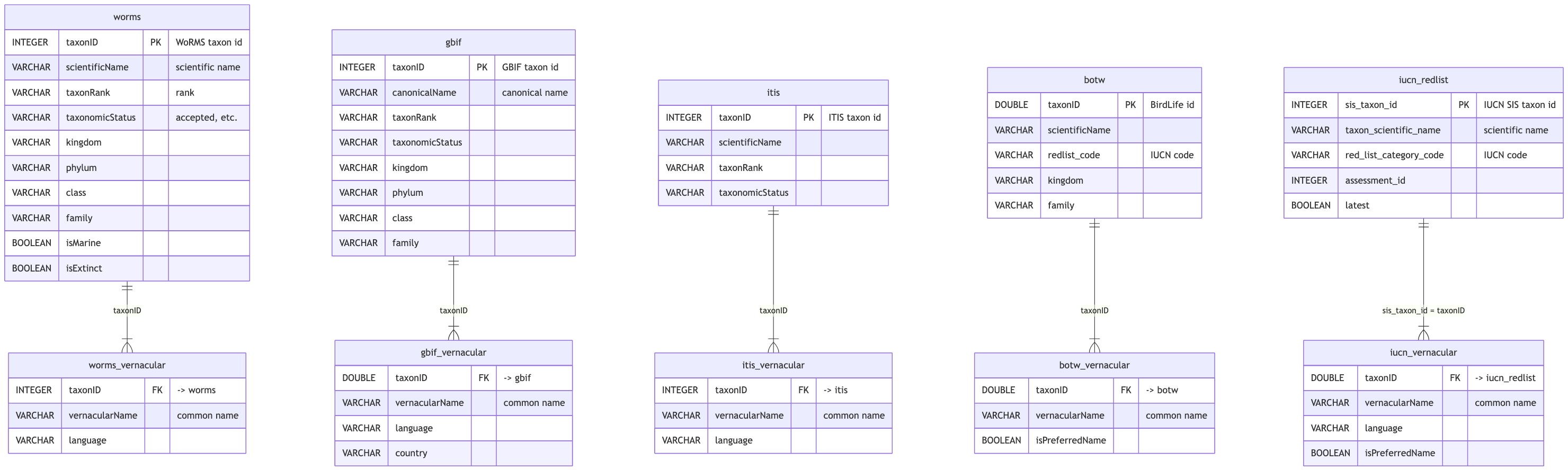
# spp.duckdb — taxonomy authority
Read-only reference of five taxonomic sources (each with a `_vernacular` common-name table).
`merge_taxon` reads WoRMS class/phylum for the taxonomy-based `sp_cat`, `isMarine`/`isExtinct` flags,
and BirdLife (`botw`) for the marine-bird determination.
```{r}
#| label: spp-changes
#| echo: false
#| output: asis
check_schema(con_spp, spp_expected, core_tables$spp, "spp.duckdb")
```
## Schema
```{mermaid}
%%| label: fig-spp-erd
%%| fig-cap: "spp.duckdb entity relationship diagram (click to zoom)"
%%| fig-width: 10
erDiagram
worms {
INTEGER taxonID PK "WoRMS AphiaID"
VARCHAR scientificName
VARCHAR class
VARCHAR phylum
VARCHAR family
BOOLEAN isMarine
BOOLEAN isExtinct
}
worms_vernacular {
INTEGER taxonID FK "-> worms"
VARCHAR vernacularName
}
botw {
DOUBLE taxonID PK "BirdLife id"
VARCHAR scientificName
VARCHAR redlist_code
VARCHAR family
}
botw_vernacular {
DOUBLE taxonID FK "-> botw"
VARCHAR vernacularName
}
gbif {
INTEGER taxonID PK "GBIF id"
VARCHAR canonicalName
VARCHAR taxonRank
VARCHAR family
}
gbif_vernacular {
DOUBLE taxonID FK "-> gbif"
VARCHAR vernacularName
}
itis {
INTEGER taxonID PK "ITIS TSN"
VARCHAR scientificName
VARCHAR taxonRank
}
itis_vernacular {
INTEGER taxonID FK "-> itis"
VARCHAR vernacularName
}
iucn_redlist {
INTEGER sis_taxon_id PK "IUCN SIS id"
VARCHAR taxon_scientific_name
VARCHAR red_list_category_code
BOOLEAN latest
}
iucn_vernacular {
DOUBLE taxonID FK "-> iucn_redlist"
VARCHAR vernacularName
}
worms ||--|{ worms_vernacular : taxonID
botw ||--|{ botw_vernacular : taxonID
gbif ||--|{ gbif_vernacular : taxonID
itis ||--|{ itis_vernacular : taxonID
iucn_redlist ||--|{ iucn_vernacular : "sis_taxon_id = taxonID"
```
## Tables
```{r}
#| label: spp-tables
#| output: asis
#| echo: false
if (!is.null(con_spp)) tables_panel(con_spp, core_tables$spp, "spp") else cat("_spp.duckdb not found locally._")
```
```{r}
#| label: cleanup
#| include: false
for (cn in list(con_serve, con_sdm, con_merge, con_spp))
if (!is.null(cn)) try(dbDisconnect(cn, shutdown = TRUE), silent = TRUE)
```