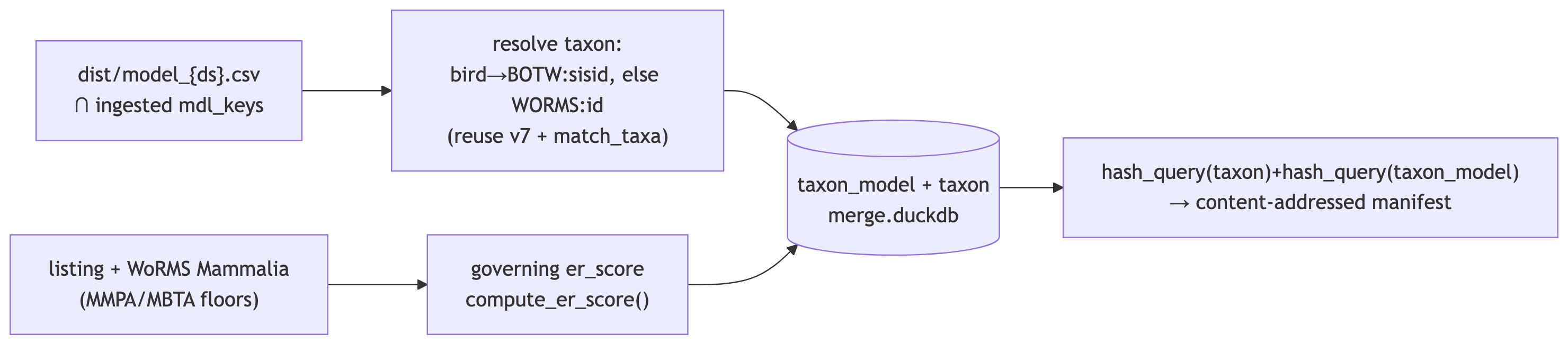
Merge prep — taxon crosswalk (each model → WORMS / BOTW taxon)
Resolve every per-dataset model to a common taxon, so models of the same species across datasets merge together. Two taxon namespaces (as in v7):
- Birds →
BOTW:{sisid}— native BirdLife id. WoRMS has poor bird coverage, so birds are NOT resolved through WoRMS. - Non-birds →
WORMS:{worms_id}— resolved fromscientific_nameby reusing v7’s taxon table (scientific_name → worms_id, cheap + already curated) andmsens::match_taxa()(ITIS→WoRMS crosswalk, exact WoRMS match, then WoRMS REST) for species v7 didn’t have.
Emits taxon (one row per taxon: id, authority, name, worms_is_marine…) and taxon_model (mdl_key ↔︎ taxon_id), keyed for merge by mdl_key_merged(authority, taxon_id) → ms_merge|WORMS:… / ms_merge|BOTW:….
1 Design
2 Setup + read all model crosswalks
Code
librarian::shelf(
DBI, dplyr, duckdb, fs, glue, here, jsonlite, logger, purrr, readr, stringr,
tibble, MarineSensitivity/msens, quiet = T)
source(here("libs/paths.R")) # dir_big_v, spp_db, ver, ver_prev, dir_big
source(here("libs/vars.R"))
options(readr.show_col_types = F)
dir_atlas <- glue("{dir_big_v}/marine-atlas")
dir_dist <- glue("{dir_atlas}/dist")
v7_sdm_db <- glue("{dir_big}/{ver_prev}/sdm.duckdb") # reuse v7 taxon resolution
manifest <- here("data/manifests/merge_models_prep.json")
dir_create(path_dir(manifest))
# model_{ds}.csv is the dataset's species crosswalk (may list MORE than were
# ingested, e.g. FWS lists all 2,196 but ingests 106) — so key off the mdl_keys that
# actually have model_cell Parquet, joined to the CSVs for scientific_name.
sci_cols <- c("scientific_name", "sci_name", "sciname", "SCIENAME", "taxa")
lookup <- dir_ls(dir_dist, glob = "*model_*.csv") |>
set_names(\(f) str_remove(path_ext_remove(path_file(f)), "^model_")) |>
imap(\(f, ds) {
m <- read_csv(f)
sci <- intersect(sci_cols, names(m))[1]
iucn <- intersect(c("category", "code", "iucn"), names(m))[1] # IUCN Red List code (rng_iucn/bl)
tibble(
mdl_key = m$mdl_key,
ds_key = ds,
scientific_name = m[[sci]],
iucn_code = if (is.na(iucn)) NA_character_ else as.character(m[[iucn]]),
sisid = if ("sisid" %in% names(m)) as.character(m$sisid) else NA_character_) }) |>
bind_rows() |> distinct(mdl_key, .keep_all = TRUE)
# read ingested mdl_keys per dataset so a mid-transfer / corrupt dataset (e.g. am
# actively rsyncing, with a truncated "no magic bytes" parquet) is skipped with a
# warning rather than crashing the whole glob — its taxa resolve on the next run.
con0 <- dbConnect(duckdb())
ds_dirs <- dir_ls(dir_dist, type = "directory", regexp = "dataset=")
ingested <- unlist(lapply(ds_dirs, \(d) tryCatch(
dbGetQuery(con0, glue("SELECT DISTINCT mdl_key FROM read_parquet('{d}/*.parquet')"))$mdl_key,
error = \(e) { log_warn("skip {path_file(d)}: {conditionMessage(e)}"); character(0) })))
dbDisconnect(con0, shutdown = TRUE)
models <- lookup |> filter(mdl_key %in% ingested)
count(models, ds_key)# A tibble: 8 × 2
ds_key n
<chr> <int>
1 am 23699
2 bl 10995
3 ca_nmfs 1
4 ch_fws 29
5 ch_nmfs 38
6 rng_fws 106
7 rng_iucn 6246
8 rng_turtle_swot_dps 63 Non-bird taxa → worms_id (reuse v7, then match_taxa)
Code
nonbird <- models |> filter(ds_key != "bl") |> distinct(scientific_name) |>
mutate(sci_clean = clean_sci_name(scientific_name), # drop (=synonym), ssp. markers
sci_binom = clean_sci_name(scientific_name, binomial = TRUE)) # Genus species fallback
# reuse v7's taxon resolution, matched on the cleaned name
con_v7 <- dbConnect(duckdb(v7_sdm_db, read_only = TRUE))
v7_tax <- tbl(con_v7, "taxon") |> filter(taxon_authority == "worms") |>
select(scientific_name, worms_id, worms_is_marine, worms_is_extinct) |> collect()
dbDisconnect(con_v7, shutdown = TRUE)
v7_tax <- v7_tax |> mutate(sci_clean = clean_sci_name(scientific_name)) |>
distinct(sci_clean, .keep_all = TRUE) |> select(-scientific_name)
resolved <- nonbird |> left_join(v7_tax, by = "sci_clean")
log_info("non-bird taxa: {nrow(resolved)} | reused from v7: {sum(!is.na(resolved$worms_id))}")
# match_taxa (ITIS->WoRMS, exact, REST) for the rest: cleaned name, then binomial fallback
match_names <- function(v) {
con_spp <- dbConnect(duckdb(spp_db, read_only = TRUE)); on.exit(dbDisconnect(con_spp, shutdown = TRUE))
match_taxa(tibble(scientific_name = unique(v)), con_spp) |>
filter(!is.na(worms_id)) |> distinct(scientific_name, .keep_all = TRUE)
}
for (col in c("sci_clean", "sci_binom")) {
todo <- resolved |> filter(is.na(worms_id)) |> pull(.data[[col]]) |> unique()
if (length(todo) == 0) next
m <- match_names(todo) |> transmute(!!col := scientific_name, wid = worms_id)
resolved <- resolved |> left_join(m, by = col) |>
mutate(worms_id = coalesce(worms_id, wid)) |> select(-wid)
}match_taxa: 727 taxa unmatched, querying WoRMS REST API...Given ~ 1 second per request and 727 unique values of `scientific_name`,
estimated time of completion: 2026-07-14 10:13:53.630752[working] (700 + 0) -> 10 -> 17 | ■■ 2%[working] (615 + 0) -> 10 -> 102 | ■■■■■ 14%[waiting] (547 + 2) -> 0 -> 180 | ■■■■■■■■ 25%Waiting 4s for rate limit ■■■■■■■■ Waiting 4s for rate limit ■■■■■■■■■■■■■■■■■■■■■■■■■■ Waiting 4s for rate limit ■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■ [waiting] (547 + 2) -> 0 -> 180 | ■■■■■■■■ 25%
[working] (477 + 24) -> 10 -> 240 | ■■■■■■■■■■■ 33%
[waiting] (415 + 26) -> 0 -> 312 | ■■■■■■■■■■■■■■ 43%
[waiting] (355 + 30) -> 0 -> 372 | ■■■■■■■■■■■■■■■■ 51%
[waiting] (294 + 33) -> 0 -> 433 | ■■■■■■■■■■■■■■■■■■■ 60%
[working] (258 + 33) -> 10 -> 459 | ■■■■■■■■■■■■■■■■■■■■ 63%
[working] (204 + 38) -> 10 -> 513 | ■■■■■■■■■■■■■■■■■■■■■■ 71%
[working] (176 + 41) -> 9 -> 542 | ■■■■■■■■■■■■■■■■■■■■■■■ 75%
[waiting] (115 + 44) -> 0 -> 612 | ■■■■■■■■■■■■■■■■■■■■■■■■■■ 84%
[working] (76 + 49) -> 10 -> 641 | ■■■■■■■■■■■■■■■■■■■■■■■■■■■ 88%
[working] (28 + 54) -> 10 -> 689 | ■■■■■■■■■■■■■■■■■■■■■■■■■■■■■ 95%
[waiting] (10 + 57) -> 0 -> 717 | ■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■ 99%
[working] (0 + 57) -> 0 -> 727 | ■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■ 100%
match_taxa: 154 taxa unmatched, querying WoRMS REST API...
Given ~ 1 second per request and 154 unique values of `scientific_name`,
estimated time of completion: 2026-07-14 10:05:12.765751
[working] (120 + 0) -> 10 -> 24 | ■■■■■■ 16%
[working] (39 + 0) -> 10 -> 105 | ■■■■■■■■■■■■■■■■■■■■■ 68%
[waiting] (5 + 2) -> 1 -> 148 | ■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■ 96%
[working] (0 + 2) -> 0 -> 154 | ■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■ 100%Code
log_info("non-bird taxa resolved: {sum(!is.na(resolved$worms_id))} of {nrow(resolved)}")4 Build taxon + taxon_model
Code
# BOTW-sisid <-> WoRMS-aphia crosswalk (birds): BirdLife is the bird authority, so BOTW:{sisid}
# is a bird's canonical taxon. Map cleaned scientific_name -> sisid so a bird's NON-bl datasets
# (FWS ranges / critical habitat, AquaMaps — which resolve to WORMS by name) merge into that SAME
# BOTW model instead of splitting the species across BOTW + WORMS (the duplicate-picker bug). Match
# on the cleaned name, with a binomial fallback for authorship/subspecies differences.
bird_xw <- models |> filter(ds_key == "bl", !is.na(sisid)) |>
transmute(sci_clean = clean_sci_name(scientific_name),
sci_binom = clean_sci_name(scientific_name, binomial = TRUE),
sisid = as.character(sisid))
# distinct maps (s_cln/s_bin names avoid colliding with models' own all-NA `sisid` column)
sisid_by_clean <- bird_xw |> distinct(sci_clean, .keep_all = TRUE) |> transmute(sci_clean, s_cln = sisid)
sisid_by_binom <- bird_xw |> distinct(sci_binom, .keep_all = TRUE) |> transmute(sci_binom, s_bin = sisid)
# non-bl models: resolve to worms by name, THEN override to the bird's BOTW:{sisid} when the
# species is a BirdLife bird (so all of a bird's datasets land on one BOTW model).
tm_nonbl <- models |> filter(ds_key != "bl") |>
mutate(sci_clean = clean_sci_name(scientific_name),
sci_binom = clean_sci_name(scientific_name, binomial = TRUE)) |>
left_join(resolved |> select(scientific_name, worms_id), by = "scientific_name") |>
left_join(sisid_by_clean, by = "sci_clean") |>
left_join(sisid_by_binom, by = "sci_binom") |>
mutate(bird_sisid = coalesce(s_cln, s_bin), # bird? (clean match, else binomial)
taxon_authority = if_else(!is.na(bird_sisid), "botw", "worms"),
taxon_id = if_else(!is.na(bird_sisid), bird_sisid, as.character(worms_id))) |>
filter(!is.na(taxon_id)) |>
transmute(mdl_key, taxon_authority, taxon_id)
# bl models -> botw taxon (native sisid)
tm_botw <- models |> filter(ds_key == "bl", !is.na(sisid)) |>
transmute(mdl_key, taxon_authority = "botw", taxon_id = as.character(sisid))
taxon_model <- bind_rows(tm_nonbl, tm_botw) |>
mutate(ms_merge_key = mdl_key_merged(taxon_authority, taxon_id))
log_info("bird crosswalk: {sum(tm_nonbl$taxon_authority=='botw')} non-bl models re-keyed BOTW (would split otherwise)")
taxon <- taxon_model |>
left_join(models |> select(mdl_key, scientific_name, iucn_code), by = "mdl_key") |>
group_by(taxon_authority, taxon_id, ms_merge_key) |>
summarise(
scientific_name = first(scientific_name),
iucn_code = first(c(iucn_code[!is.na(iucn_code)], NA_character_)), # IUCN category if any model has it
n_models = n_distinct(mdl_key),
n_datasets = n_distinct(str_extract(mdl_key, "^[^|]+")), .groups = "drop") |>
left_join(resolved |> select(scientific_name, worms_is_marine, worms_is_extinct),
by = "scientific_name")
con <- dbConnect(duckdb(glue("{dir_atlas}/merge.duckdb")))
dbWriteTable(con, "taxon_model", taxon_model, overwrite = TRUE)
dbWriteTable(con, "taxon", taxon, overwrite = TRUE)
dbDisconnect(con, shutdown = TRUE)
glue("{nrow(taxon)} taxa ({sum(taxon$taxon_authority=='worms')} WORMS + ",
"{sum(taxon$taxon_authority=='botw')} BOTW) from {nrow(taxon_model)} models")37051 taxa (26056 WORMS + 10995 BOTW) from 41015 models5 Governing extinction-risk score (er_score)
Each ingest layer holds the ER as its own dataset knew it — IUCN category for rng_iucn/BOTW, FWS status for rng_fws/ch_fws, NMFS for SWOT/critical-habitat — each via msens::compute_er_score(). Those source values can be out of date (an older IUCN category) or out of context (a US national NMFS/FWS listing, or an MMPA/MBTA floor, that should override the IUCN international score). So here we compute each taxon’s governing er_score: the most-protective across its datasets, US national overriding IUCN, with MMPA/MBTA floors — again via compute_er_score(). merge_models applies this to the range-cell values (the fitting point where a species’ presence should carry its governing ER, not the raw source code); v7 followed the same model. Needs the listing table (ingest_listings).
The two statutory floors differ in how they are assigned:
- MMPA (
is_mmpa, +20 floor) — the Marine Mammal Protection Act protects all marine mammals, so we assign it by taxonomy: every WoRMS class Mammalia taxon (matched by AphiaID =taxon_id), not the incomplete NMFS directory (which lists only ~70 managed stocks vs 137 marine-mammal taxa here). - MBTA (
is_mbta, +10 floor) — the Migratory Bird Treaty Act protects only migratory birds native to the US / US territories, an explicit species list, so we assign it from the authoritative FWS CFR 50 §10.13 list (ingest_listings, matched byclean_sci_name). It is not all Aves — a bird absent from 10.13 (e.g. a non-US / non-migratory species) gets no MBTA floor. Most 10.13 birds are terrestrial; the marine-relevance filter (score_zones) is what keeps terrestrial birds out of the score, while this floor only sets the value of those that remain.
Code
con <- dbConnect(duckdb(glue("{dir_atlas}/merge.duckdb")))
tx <- dbGetQuery(con, "SELECT ms_merge_key, taxon_authority, taxon_id, scientific_name, iucn_code FROM taxon") |>
mutate(sci = clean_sci_name(scientific_name))
# US ESA + MBTA + BCC stay species-specific from the listing. is_mbta is the FWS CFR
# 50 §10.13 list (migratory birds native to the US) — an explicit species list, NOT all
# Aves; a bird absent from 10.13 gets no MBTA floor.
if ("listing" %in% dbListTables(con)) {
tx <- tx |> left_join(dbGetQuery(con, "SELECT sci, nmfs_esa, fws_esa, is_mbta, is_bcc FROM listing"), by = "sci")
} else {
log_warn("no listing table — run ingest_listings; ESA/MBTA fall back to IUCN only")
tx <- tx |> mutate(nmfs_esa = NA, fws_esa = NA, is_mbta = FALSE, is_bcc = FALSE)
}
# MMPA is assigned by TAXONOMY (all marine mammals are protected): every WoRMS class
# Mammalia taxon (AphiaID = taxon_id), more complete than the ~70-stock NMFS directory.
con_spp <- dbConnect(duckdb(spp_db, read_only = TRUE))
mammalia <- dbGetQuery(con_spp, "SELECT DISTINCT CAST(scientificNameID AS VARCHAR) aphia FROM worms WHERE class='Mammalia'")$aphia
dbDisconnect(con_spp, shutdown = TRUE)
esa_rank <- function(x) match(coalesce(x, "LC"), c("LC", "TN", "EN"))
tx <- tx |> mutate(
is_bcc = coalesce(is_bcc, FALSE),
is_mmpa = taxon_authority == "worms" & taxon_id %in% mammalia, # MMPA: all marine mammals
is_mbta = coalesce(is_mbta, FALSE) & taxon_authority == "botw", # MBTA: CFR 10.13, birds only
us_code = c("LC", "TN", "EN")[pmax(esa_rank(nmfs_esa), esa_rank(fws_esa))],
is_us = us_code != "LC" | is_mmpa | is_mbta,
extrisk_code = case_when(
is_us & is_mmpa ~ paste0("NMFS:", us_code), # US national + MMPA overrides IUCN
is_us ~ paste0("FWS:", us_code),
iucn_code %in% c("CR","EN","VU","NT","LC","DD") ~ paste0("IUCN:", iucn_code),
TRUE ~ NA_character_))
tx$er_score <- 1L
ok <- !is.na(tx$extrisk_code)
tx$er_score[ok] <- compute_er_score(tx$extrisk_code[ok], is_mmpa = tx$is_mmpa[ok], is_mbta = tx$is_mbta[ok])
dbWriteTable(con, "taxon_er",
tx |> select(ms_merge_key, extrisk_code, er_score, is_mmpa, is_mbta, is_bcc), overwrite = TRUE)
dbExecute(con, "CREATE OR REPLACE TABLE taxon AS
SELECT t.*, e.extrisk_code, e.er_score, e.is_mmpa, e.is_mbta, e.is_bcc
FROM taxon t LEFT JOIN taxon_er e USING (ms_merge_key)")[1] 37051Code
log_info("taxonomy floors: {sum(tx$is_mmpa)} Mammalia (MMPA), {sum(tx$is_mbta)} Aves (MBTA); {sum(tx$is_us)} US-listed of {nrow(tx)}")
dbDisconnect(con, shutdown = TRUE)6 Outputs + manifest
Code
# content-addressed: fingerprint of the taxon + taxon_model output tables (reopen
# read-only since the er_score chunk already disconnected)
con <- dbConnect(duckdb(glue("{dir_atlas}/merge.duckdb"), read_only = TRUE))
h <- paste0(msens::hash_query(con, "taxon"), msens::hash_query(con, "taxon_model"))
dbDisconnect(con, shutdown = TRUE)
smry <- tibble(n_taxa = nrow(taxon), n_models = nrow(taxon_model),
n_worms = sum(taxon$taxon_authority == "worms"),
n_botw = sum(taxon$taxon_authority == "botw"))
msens::report_table(smry, caption = "merge_models_prep: taxa + models")| n_taxa | n_models | n_worms | n_botw |
|---|---|---|---|
| 37051 | 41015 | 26056 | 10995 |
Code
msens::write_manifest(
manifest, target = "merge_models_prep", content_hash = h,
stats = list(ver = ver, n_models = smry$n_models, n_taxa = smry$n_taxa,
n_worms = smry$n_worms, n_botw = smry$n_botw),
force = msens::force_target("merge_models_prep"))