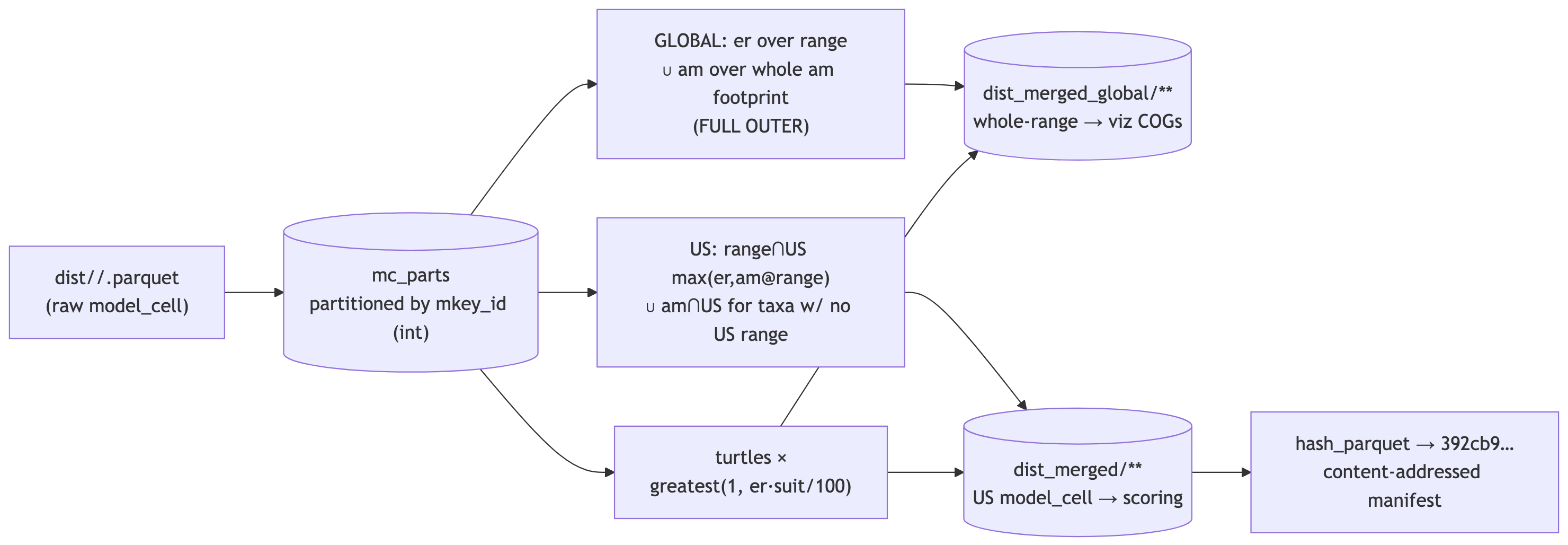
Merge models — combine each taxon’s models per cell (max-merge + range mask; turtles ×)
Combine every taxon’s per-dataset models into one merged model_cell (mdl_key = ms_merge|WORMS:… / BOTW:…), reproducing v7’s logic at the v8 global scale (~13B am cell-rows), so it must stream + spill rather than materialize:
- Default = max-merge, masked to the range footprint. A taxon’s merged cells are its range cells if it has any (
rng_*/ch_*/ca_*/bl), else its fullamfootprint; at each kept cellval = maxacross the taxon’s models. Streamed straight to Parquet via a singleCOPY(one grouped scan), so no 13B-row table. - Turtles = multiplicative.
val = pmax(1, round(er × suit / 100))with critical-habitat max-override — computed from only the 6 turtle taxa’s files (never a full am scan, which is what OOM’d the naive version).
1 Design
2 Setup
Code
librarian::shelf(DBI, dplyr, duckdb, fs, glue, here, jsonlite, logger, quiet = T)
source(here("libs/paths.R"))
source(here("libs/vars.R"))
dir_atlas <- glue("{dir_big_v}/marine-atlas")
dir_dist <- glue("{dir_atlas}/dist")
merge_db <- glue("{dir_atlas}/merge.duckdb")
# ONE global merge → two surfaces from the same data:
dir_global <- glue("{dir_atlas}/dist_merged_global/dataset=ms_merge") # whole-range (land+ocean) → viz COGs
dir_merged <- glue("{dir_atlas}/dist_merged/dataset=ms_merge") # US-study-area trim → model_cell scoring
manifest <- here("data/manifests/merge_models.json")
dir_create(path_dir(manifest))
stopifnot("run merge_models_prep first" = file_exists(merge_db),
"run build_cell_grid first" = file_exists(sdm_db))
suit_ds <- "am"
turtle_ds <- "rng_turtle_swot_dps"
ch_ds <- c("ch_nmfs", "ch_fws", "ca_nmfs")
ch_sql <- paste(sprintf("'%s'", ch_ds), collapse = ",")
con <- dbConnect(duckdb(merge_db))
# spill config so the multi-billion-row aggregation goes out-of-core instead of OOM. Keep the
# memory limit WELL under physical RAM (other processes — browser, renders — compete), cap threads
# (each parallel aggregate/join holds hash tables), and allow a large temp spill.
mem_gb <- tryCatch(
max(4, min(8, floor(as.numeric(system("sysctl -n hw.memsize", intern = TRUE)) / 1e9 * 0.3))),
error = function(e) 6)
dbExecute(con, glue("PRAGMA memory_limit='{mem_gb}GB'"))[1] 0Code
dbExecute(con, glue("PRAGMA temp_directory='{dir_atlas}/duckdb_tmp'"))[1] 0Code
dbExecute(con, glue("PRAGMA max_temp_directory_size='400GB'"))[1] 0Code
dbExecute(con, glue("PRAGMA threads={min(4L, max(1L, parallel::detectCores() - 1L))}"))[1] 0Code
# ONE GLOBAL merge, two outputs from the same data: the whole-range merged surface
# (dist_merged_global → viz COGs) and, trimmed to the US study area, the model_cell scoring surface
# (dist_merged). am is DENSE (~13B rows, 50G) so it is NEVER grouped: range cells (non-am) are
# materialized once + indexed (mc_range, ~3B); am is a VIEW (mc_am) probed only at the sparse range
# cells (both/turtle) or streamed for am-only taxa. us_cells is applied AFTER the global merge.
dbExecute(con, glue("ATTACH '{sdm_db}' AS grid (READ_ONLY)"))[1] 0Code
dbExecute(con, "CREATE OR REPLACE TABLE us_cells AS SELECT cell_id FROM grid.cell WHERE in_usa")[1] 634208Code
n_us <- dbGetQuery(con, "SELECT count(*) n FROM us_cells")$n
# PARTITION the full merge input (ranges + am, ~16B rows) by ms_merge_key — a STREAMING write that
# never holds it in memory. The merge below then reads one BATCH of taxa at a time via
# `ms_merge_key IN (...)`, so DuckDB partition-prunes to just those taxa's slices: bounded memory,
# no OOM (a bulk join over the ~3B `both` range cells otherwise blows the memory limit).
try(dbExecute(con, "DROP TABLE IF EXISTS model_cell_all"), silent = TRUE)[1] 0Code
try(dbExecute(con, "DROP TABLE IF EXISTS mc_range"), silent = TRUE)[1] 0Code
# ms_merge_key holds '|' and ':' which break hive partition PATHS (…/mc_parts/ms_merge_key=ms_merge|…
# can't be parsed back). Partition on an INTEGER surrogate mkey_id — exactly what the serve surface
# does with mdl_id. ms_merge_key stays a stored column.
dbExecute(con, "CREATE OR REPLACE TABLE mkey_map AS
SELECT ms_merge_key, CAST(dense_rank() OVER (ORDER BY ms_merge_key) AS INTEGER) AS mkey_id
FROM (SELECT DISTINCT ms_merge_key FROM taxon_model)")[1] 37051Code
dir_parts <- glue("{dir_atlas}/mc_parts")
# the partition write is the ~40-min step; skip it if the input already exists (REDO_MC_PARTS=1 forces)
if (!dir_exists(dir_parts) || nzchar(Sys.getenv("REDO_MC_PARTS"))) {
if (dir_exists(dir_parts)) dir_delete(dir_parts)
dbExecute(con, "SET partitioned_write_max_open_files=512")
msens::copy_atlas_parquet(con, glue(
"SELECT mm.mkey_id, tm.ms_merge_key, split_part(mc.mdl_key,'|',1) AS ds_key, mc.cell_id, mc.val
FROM read_parquet('{dir_dist}/*/*.parquet') mc JOIN taxon_model tm USING (mdl_key)
JOIN mkey_map mm USING (ms_merge_key)"),
dir_parts, partition_by = "mkey_id")
} else log_info("mc_parts exists — skipping repartition (REDO_MC_PARTS=1 to force)")
# VIEW over the partitioned input — EVERY query below filters mkey_id (integer) so reads are pruned
dbExecute(con, glue("CREATE OR REPLACE VIEW mc_parts AS
SELECT mkey_id, ms_merge_key, ds_key, cell_id, val
FROM read_parquet('{dir_parts}/**/*.parquet', hive_partitioning = true)"))[1] 0Code
all_ids <- dbGetQuery(con, "SELECT mkey_id FROM mkey_map ORDER BY mkey_id")$mkey_id
log_info("partitioned merge input: {length(all_ids)} taxa by mkey_id; US trim {n_us} in_usa cells; memory_limit={mem_gb}GB")3 Turtle taxa (multiplicative, from only their files)
Code
turtle_keys <- dbGetQuery(con, glue(
"SELECT DISTINCT ms_merge_key FROM taxon_model WHERE mdl_key LIKE '{turtle_ds}|%'"))$ms_merge_key
tkeys_sql <- paste(sprintf("'%s'", turtle_keys), collapse = ",")
turtle_ids <- dbGetQuery(con, glue("SELECT mkey_id FROM mkey_map WHERE ms_merge_key IN ({tkeys_sql})"))$mkey_id
tids_sql <- paste(turtle_ids, collapse = ",")
log_info("{length(turtle_keys)} turtle taxa (multiplicative merge)")
# whole-range (global) multiplicative turtle merge, reading ONLY the turtle taxa's partitions
# (mkey_id IN (...) → partition-pruned, tiny). Rule = msens::turtle_sql() (unit-tested).
dbExecute(con, glue("CREATE OR REPLACE TABLE turtle_src AS
SELECT ms_merge_key, ds_key, cell_id, val FROM mc_parts WHERE mkey_id IN ({tids_sql})"))[1] 150301050Code
dbExecute(con, paste("CREATE OR REPLACE TABLE mc_turtle AS",
msens::turtle_sql(turtle_ds, suit_ds, ch_ds, src = "turtle_src")))[1] 46304118Code
log_info("mc_turtle (whole-range): {dbGetQuery(con, 'SELECT count(*) n FROM mc_turtle')$n} cells")4 Max-merge (masked) + turtles → streamed to Parquet
Code
# taxon flags from the crosswalk (no cell scan)
dbExecute(con, "CREATE OR REPLACE TABLE taxon_flags AS
SELECT ms_merge_key,
bool_or(split_part(mdl_key,'|',1)='am') AS has_am,
bool_or(split_part(mdl_key,'|',1)<>'am') AS has_range
FROM taxon_model GROUP BY ms_merge_key")[1] 37051Code
# skip the ~2h merge write when the surfaces already exist — re-renders (e.g. to refresh the HTML)
# reuse them; the summaries + hash below read the on-disk parquet either way. REDO_MERGE=1 forces.
redo_merge <- nzchar(Sys.getenv("REDO_MERGE"))
have_merged <- dir_exists(dir_merged) && dir_exists(dir_global) &&
length(dir_ls(dir_merged, glob = "*.parquet", recurse = TRUE)) > 0
if (redo_merge || !have_merged) {
if (dir_exists(dir_global)) dir_delete(dir_global); dir_create(dir_global)
if (dir_exists(dir_merged)) dir_delete(dir_merged); dir_create(dir_merged)
# turtles (done) → whole-range global + US-trim
msens::copy_atlas_parquet(con, "SELECT mdl_key, cell_id, val FROM mc_turtle",
glue("{dir_global}/turtle"), per_thread = TRUE)
msens::copy_atlas_parquet(con,
"SELECT mt.mdl_key, mt.cell_id, mt.val FROM mc_turtle mt JOIN us_cells u ON mt.cell_id = u.cell_id",
glue("{dir_merged}/turtle"), per_thread = TRUE)
# BATCH the rest so each merge reads ONLY its taxa's pruned partitions (bounded memory). Two surfaces
# come off the same batch (see glob_global_sql / us_sql below): the GLOBAL viz surface = am ∪ range
# (whole-range merged model, FULL OUTER); the US SCORING surface = v7-faithful US-boundary-aware rule
# (range footprint ∩ US valued max(er, am-at-range), UNION am ∩ US for taxa with no range in the US).
# The US rule keeps the 750 am-in-US taxa whose IUCN range lies wholly outside the US (silently
# dropped before) yet still masks am to the range for taxa that DO have US range — so it reproduces
# the prior US merge exactly and is NOT a plain trim of the global surface.
# am-only taxa have no range footprint → omitted from the GLOBAL surface (reuse am COGs), but their
# US am cells go into the scoring surface via the am-only UNION branch.
# Batch by CELL COUNT, not taxa count: a few taxa have global ranges (10-25M cells each), so a
# fixed 150-taxa batch OOMs. Pack taxa until ~40M cells/batch (a single taxon maxes at the 25.9M
# grid, so it always fits one batch). Counts come from parquet metadata (fast, no data scan).
nonturtle <- setdiff(all_ids, turtle_ids)
cnt <- dbGetQuery(con, "SELECT mkey_id, count(*) AS n FROM mc_parts GROUP BY mkey_id")
cnt <- cnt[cnt$mkey_id %in% nonturtle, ]; cnt <- cnt[order(cnt$mkey_id), ]
batches <- unname(split(cnt$mkey_id, cumsum(as.numeric(cnt$n)) %/% 40e6))
log_info("merging {length(nonturtle)} non-turtle taxa in {length(batches)} cell-bounded batches (~40M cells each)")
# The merge RULES are msens::merge_sql() — the SINGLE SOURCE OF TRUTH, unit-tested in
# msens/tests/testthat/test-merge.R (every taxon category asserted). Two surfaces from the same batch:
# GLOBAL viz ($global) = am ∪ range (FULL OUTER, whole-range merged model) → COGs.
# US scoring ($us) = v7-faithful, IUCN-CONSTRAINED: range∩US max(er, am-at-range) [am BEYOND the
# range is MASKED] UNION raw am∩US for TRUE am-only taxa (global has_range=FALSE). A species whose
# IUCN range lies WHOLLY outside the US gets NO US presence — the iucn_range_outside_us_eez
# exclusion (e.g. Sotalia guianensis). See ?msens::merge_sql for the full rationale.
msq <- msens::merge_sql()
glob_global_sql <- msq$global
us_sql <- msq$us
for (i in seq_along(batches)) {
bk <- paste(batches[[i]], collapse = ",") # integer mkey_ids → partition-pruned
dbExecute(con, glue("CREATE OR REPLACE TABLE b AS
SELECT ms_merge_key, ds_key, cell_id, val FROM mc_parts WHERE mkey_id IN ({bk})"))
dbExecute(con, msq$b_range) # range footprint (non-am) valued by governing er_score
dbExecute(con, msq$b_am_all) # (global) am over WHOLE am footprint of has_range taxa → am∪range
dbExecute(con, msq$b_am_rng) # (US) am AT range cells only → range-footprint max(er, am-at-range)
msens::copy_atlas_parquet(con, glob_global_sql, glue("{dir_global}/b{i}"), per_thread = TRUE)
msens::copy_atlas_parquet(con, us_sql, glue("{dir_merged}/b{i}"), per_thread = TRUE)
if (i %% 10 == 0) log_info(" batch {i}/{length(batches)}")
}
dbExecute(con, "DROP TABLE IF EXISTS b; DROP TABLE IF EXISTS b_range;
DROP TABLE IF EXISTS b_am_all; DROP TABLE IF EXISTS b_am_rng")
} else log_info("dist_merged + dist_merged_global exist — skipping merge write (REDO_MERGE=1 to force)")
# summaries: global (viz) + US (scoring); fingerprint the US scoring surface (the tracked output)
smry_g <- dbGetQuery(con, glue("SELECT count(DISTINCT mdl_key) n_taxa, count(*) n_cells,
min(val) v_min, max(val) v_max FROM read_parquet('{dir_global}/**/*.parquet')"))
smry <- dbGetQuery(con, glue("SELECT count(DISTINCT mdl_key) n_taxa, count(*) n_cells,
min(val) v_min, max(val) v_max FROM read_parquet('{dir_merged}/**/*.parquet')"))
h <- msens::hash_parquet(glue("{dir_merged}/**/*.parquet"), con)
msens::report_table(smry_g, caption = "GLOBAL whole-range merged surface (ms_merge) → viz COGs")| n_taxa | n_cells | v_min | v_max |
|---|---|---|---|
| 17129 | 4088690681 | 1 | 100 |
Code
msens::report_table(smry, caption = "US-trimmed merged model_cell (ms_merge) → scoring")| n_taxa | n_cells | v_min | v_max |
|---|---|---|---|
| 17763 | 580568326 | 1 | 100 |
Code
dbDisconnect(con, shutdown = TRUE)5 Manifest
Code
# content-addressed: fingerprint of the US scoring surface (the tracked target output); the global
# viz surface (dist_merged_global) is recorded in stats but doesn't drive the hash.
msens::write_manifest(
manifest, target = "merge_models", content_hash = h,
stats = list(ver = ver, n_taxa = smry$n_taxa, n_cells = smry$n_cells,
v_min = smry$v_min, v_max = smry$v_max,
n_taxa_global = smry_g$n_taxa, n_cells_global = smry_g$n_cells),
force = msens::force_target("merge_models"))