---
title: "Investigate Rice's Whale DB Corruption"
subtitle: "Diagnose spatial data corruption in model_cell for Rice's whale and other species"
format:
html:
code-fold: true
code-tools: true
editor_options:
chunk_output_type: console
---
## Overview
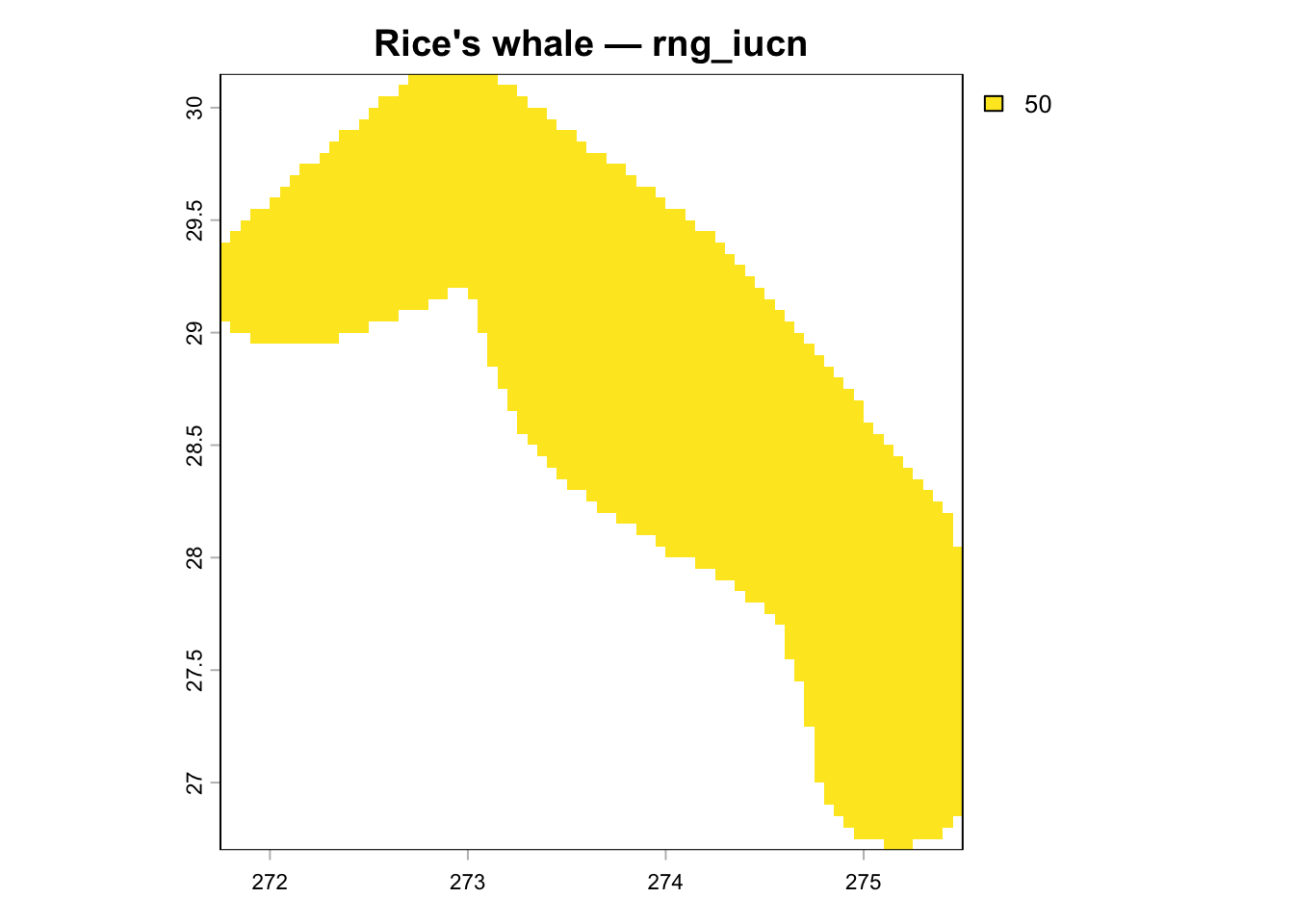
Rice's whale (taxon_id=1576133) rng_iucn shows only ~1,969 cells (identical to
ca_nmfs), but the correct IUCN polygon covers ~49,500 km² and should produce
~3,000-4,000 cells. The Critical Habitat (ch_nmfs) has 2,677 cells — more than
the supposedly-larger IUCN range, which is backwards. This notebook diagnoses
when/how the corruption occurred by comparing database backups and raw source
files.
## Setup
```{r}
#| label: setup
librarian::shelf(
DBI,
dplyr,
duckdb,
glue,
here,
leaflet,
purrr,
sf,
terra,
tibble,
quiet = T
)
source(here("libs/paths.R"))
cell_tif <- glue("{dir_derived}/r_bio-oracle_planarea.tif")
r_cell <- rast(cell_tif, lyrs = "cell_id")
```
## Connect to Database Versions
```{r}
#| label: connect_dbs
dir_archive <- glue("{dir_big}/archive_2026-02-12")
# define database paths (read-only)
db_paths <- c(
"Jan 29" = glue("{dir_archive}/sdm_2026-01-29.duckdb"),
"Feb 10" = glue("{dir_archive}/sdm_2026-02-10.duckdb"),
"Feb 12 (v3 backup)" = glue("{dir_archive}/sdm_v3_2026-02-12.duckdb"),
"v3 current" = glue("{dir_big}/v3/sdm.duckdb"),
"v3b" = glue("{dir_big}/v3b/sdm.duckdb")
)
# filter to paths that actually exist
db_paths <- db_paths[file.exists(db_paths)]
message(glue(
"Found {length(db_paths)} databases: {paste(names(db_paths), collapse = ', ')}"
))
# open read-only connections
cons <- map(db_paths, \(p) dbConnect(duckdb(dbdir = p, read_only = TRUE)))
names(cons) <- names(db_paths)
```
## Rice's Whale model_cell Comparison Across DBs
```{r}
#| label: ricei_comparison
taxon_id_ricei <- 1576133L
d_compare <- map_dfr(names(cons), \(db_name) {
con <- cons[[db_name]]
# check if taxon_model table exists
if (!dbExistsTable(con, "taxon_model")) {
return(tibble())
}
tryCatch(
{
tbl(con, "taxon_model") |>
filter(taxon_id == !!taxon_id_ricei) |>
select(taxon_id, mdl_seq) |>
inner_join(
tbl(con, "model") |> select(mdl_seq, ds_key),
by = "mdl_seq"
) |>
inner_join(
tbl(con, "model_cell") |>
group_by(mdl_seq) |>
summarize(
n_cells = n(),
v_min = min(value, na.rm = TRUE),
v_max = max(value, na.rm = TRUE),
.groups = "drop"
),
by = "mdl_seq"
) |>
collect() |>
mutate(db_version = db_name)
},
error = \(e) {
message(glue("Error querying {db_name}: {e$message}"))
tibble()
}
)
})
if (nrow(d_compare) > 0) {
d_compare |>
select(db_version, ds_key, mdl_seq, n_cells, v_min, v_max) |>
arrange(db_version, ds_key) |>
knitr::kable(caption = "Rice's whale model_cell summary across DB versions")
} else {
message("No data found for Rice's whale in any database")
}
```
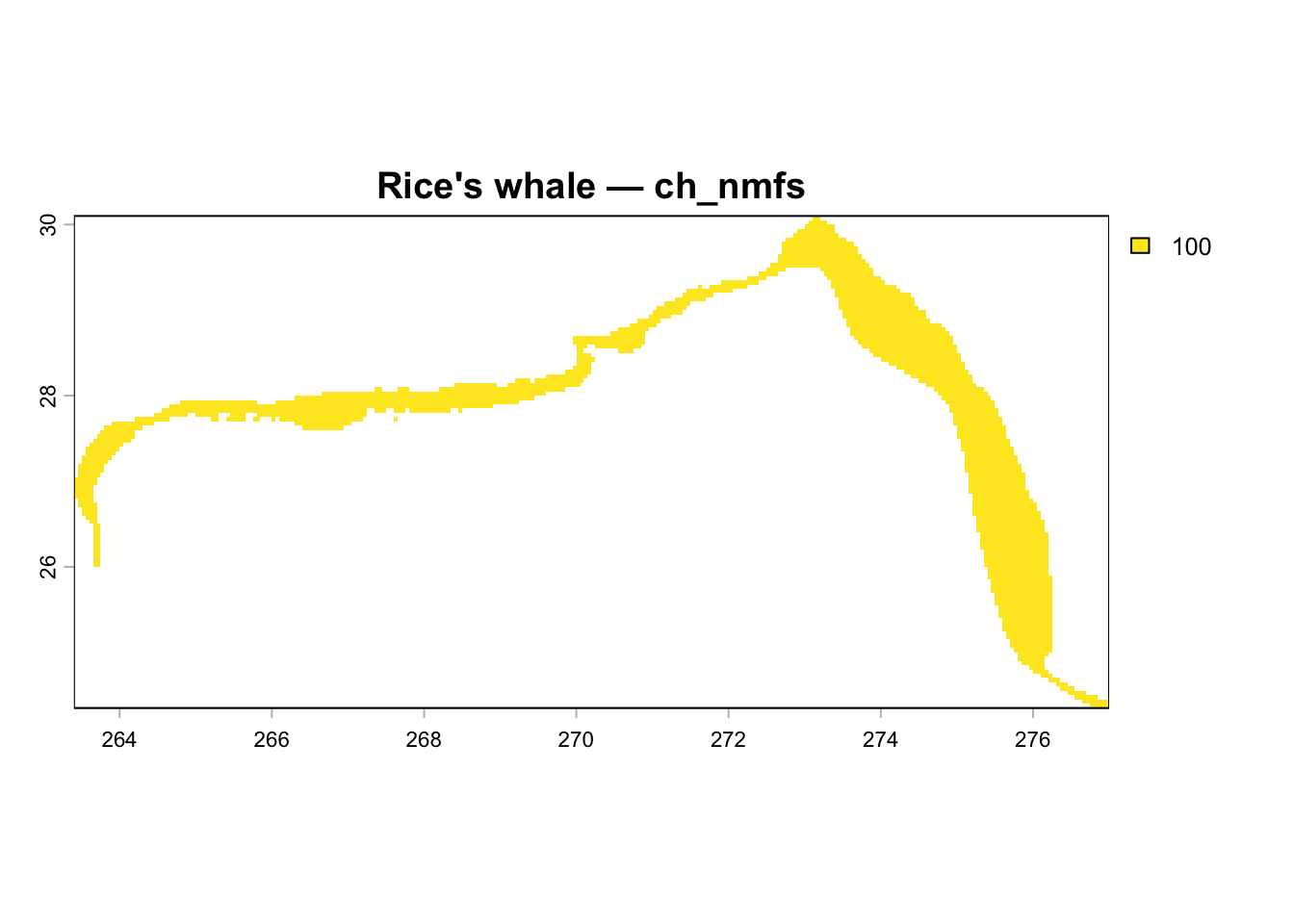
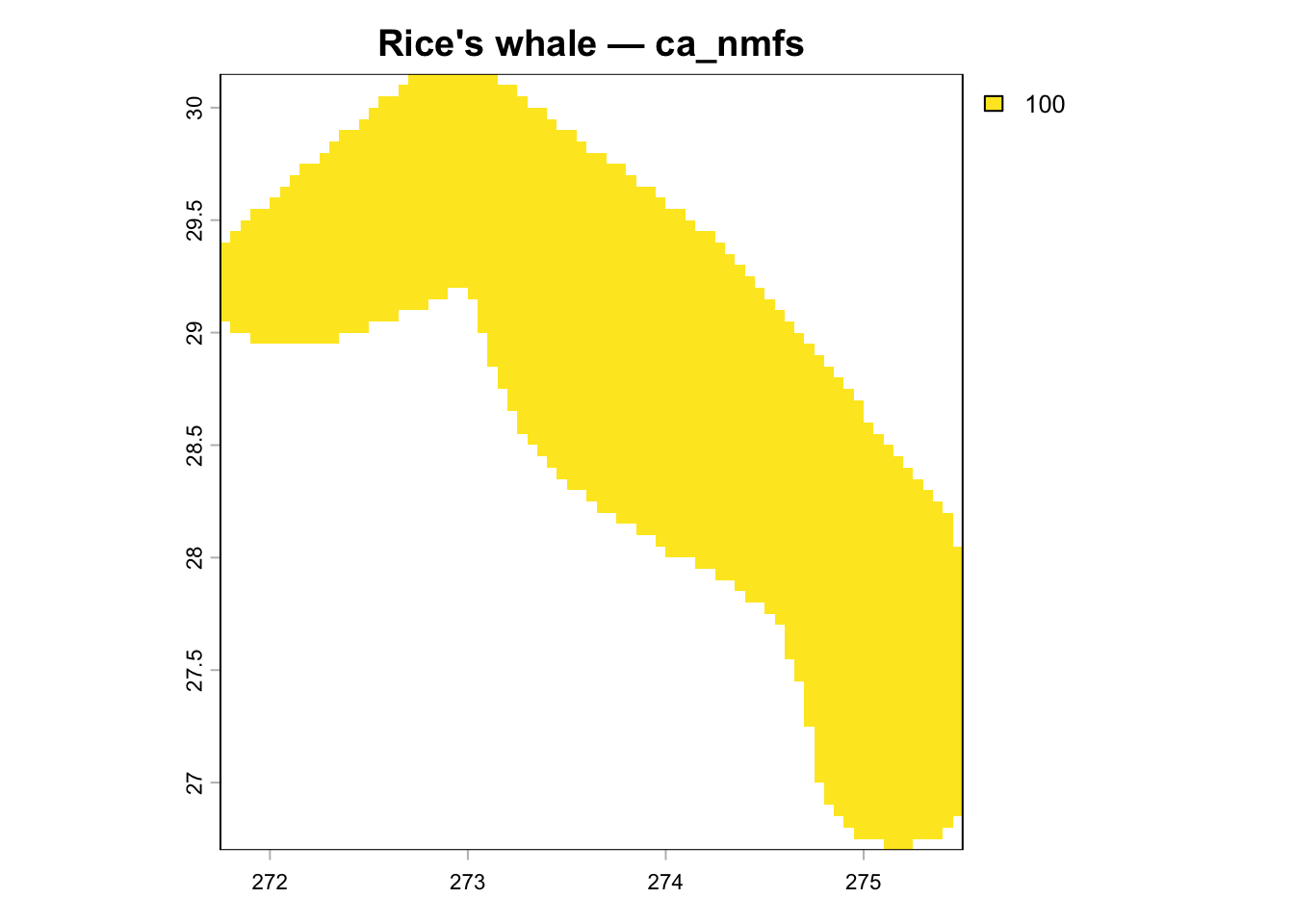
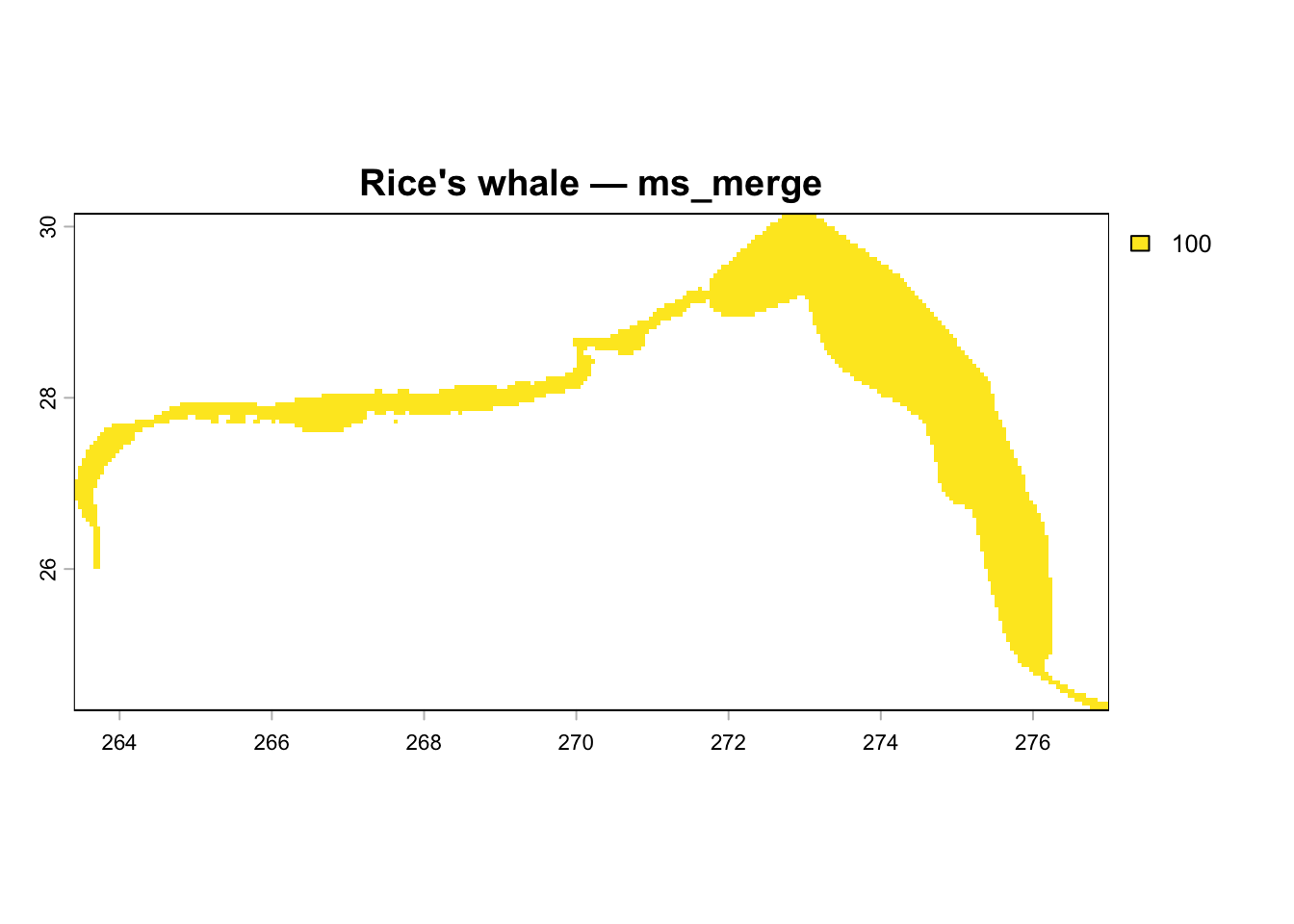
## Visualize Rice's Whale Spatial Layers
Map cell_id values back to raster for each layer from the current v3 database.
```{r}
#| label: ricei_viz
con_v3 <- cons[["v3 current"]]
# get all Rice's whale layers
d_layers <- tbl(con_v3, "taxon_model") |>
filter(taxon_id == !!taxon_id_ricei) |>
select(taxon_id, mdl_seq) |>
inner_join(
tbl(con_v3, "model") |> select(mdl_seq, ds_key),
by = "mdl_seq"
) |>
collect()
# rasterize each layer
r_layers <- map(1:nrow(d_layers), \(i) {
lyr <- d_layers[i, ]
d_mc <- tbl(con_v3, "model_cell") |>
filter(mdl_seq == !!lyr$mdl_seq) |>
collect()
r <- init(r_cell[[1]], NA)
r[d_mc$cell_id] <- d_mc$value
names(r) <- lyr$ds_key
r
})
names(r_layers) <- d_layers$ds_key
# display each layer
for (nm in names(r_layers)) {
r <- trim(r_layers[[nm]])
message(glue("{nm}: {ncell(r[!is.na(r)])} non-NA cells"))
plot(r, main = glue("Rice's whale — {nm}"))
}
```
### Interactive map comparison
```{r}
#| label: ricei_leaflet
# combine all layers into a single stack for comparison
r_all <- map(r_layers, \(r) trim(r)) |>
purrr::reduce(\(a, b) {
# extend to common extent
e <- ext(
min(ext(a)[1], ext(b)[1]),
max(ext(a)[2], ext(b)[2]),
min(ext(a)[3], ext(b)[3]),
max(ext(a)[4], ext(b)[4])
)
extend(a, e) |>
c(extend(b, e))
})
# plot the first available layer interactively
if (length(r_layers) > 0) {
r_plot <- trim(r_layers[[1]])
plet(r_plot, main = names(r_layers)[1])
}
```
## Compare DB Cells to Raw Source Polygons
### Load raw shapefiles
```{r}
#| label: load_raw_polys
# iucn range polygon
iucn_shp <- glue(
"{dir_raw}/iucnredlist.org/MAMMALS_MARINE_ONLY/MAMMALS_MARINE_ONLY.shp"
)
if (file.exists(iucn_shp)) {
sf_iucn <- st_read(iucn_shp, quiet = TRUE) |>
filter(sci_name == "Balaenoptera ricei")
message(glue(
"IUCN polygon: {nrow(sf_iucn)} features, ",
"area = {round(sum(st_area(sf_iucn)) / 1e6)} km²"
))
} else {
message(glue("IUCN shapefile not found: {iucn_shp}"))
sf_iucn <- NULL
}
# nmfs core area polygon
core_dir <- glue(
"{dir_raw}/fisheries.noaa.gov/core-areas/",
"shapefile_Rices_whale_core_distribution_area_Jun19_SERO"
)
core_shp <- glue(
"{core_dir}/",
"shapefile_Rices_whale_core_distribution_area_Jun19_SERO.shp"
)
if (file.exists(core_shp)) {
sf_core <- st_read(core_shp, quiet = TRUE)
message(glue(
"NMFS Core Area: {nrow(sf_core)} features, ",
"area = {round(sum(st_area(sf_core)) / 1e6)} km²"
))
} else {
message(glue("Core area shapefile not found: {core_shp}"))
sf_core <- NULL
}
# critical habitat from gdb
ch_gdb <- glue(
"{dir_raw}/fisheries.noaa.gov/",
"NMFS_ESA_Critical_Habitat_20230505_2025-07-17.gdb"
)
if (file.exists(ch_gdb)) {
# list layers to find Rice's whale critical habitat
lyrs <- st_layers(ch_gdb)
message(glue("GDB layers: {paste(lyrs$name, collapse = ', ')}"))
# try to find the layer containing Rice's whale
# (user may need to adjust layer name)
ricei_lyr <- lyrs$name[grepl(
"Rice|Bryde|Balaenoptera",
lyrs$name,
ignore.case = TRUE
)]
if (length(ricei_lyr) > 0) {
sf_ch <- st_read(ch_gdb, layer = ricei_lyr[1], quiet = TRUE)
message(glue("CH layer '{ricei_lyr[1]}': {nrow(sf_ch)} features"))
} else {
message("No Rice's whale layer found in GDB — listing all layers above")
sf_ch <- NULL
}
} else {
message(glue("Critical Habitat GDB not found: {ch_gdb}"))
sf_ch <- NULL
}
```
### Overlay raw polygons on rasterized cells
```{r}
#| label: overlay_comparison
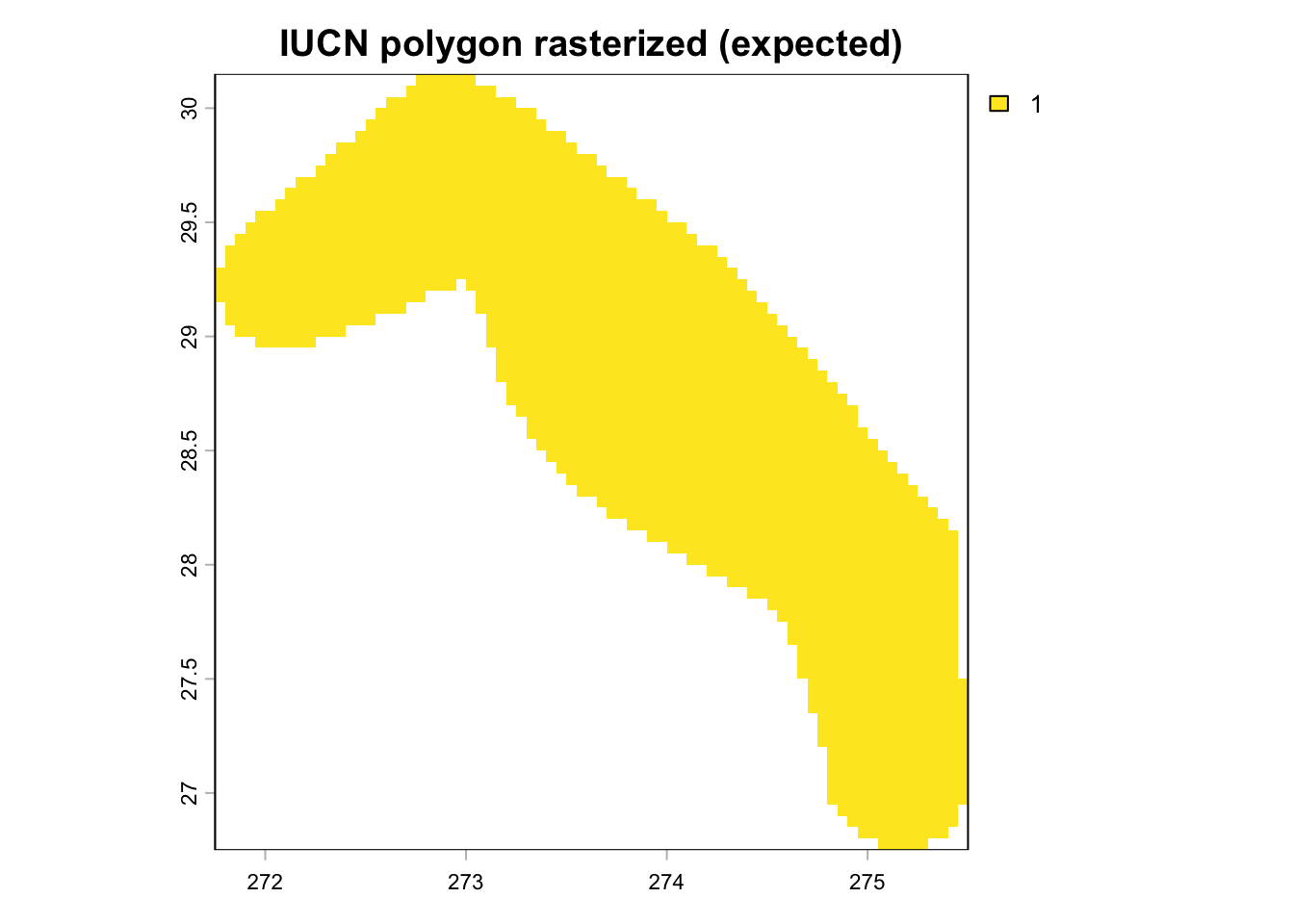
# rasterize IUCN polygon using the same grid as the DB
# note: r_cell uses 0-360 longitude; polygon uses -180-180, so shift by +360
if (!is.null(sf_iucn)) {
v_iucn <- vect(sf_iucn) |> shift(dx = 360)
r_iucn_poly <- rasterize(v_iucn, r_cell, field = 1, background = NA)
n_expected <- sum(!is.na(values(r_iucn_poly)))
message(glue(
"Expected rng_iucn cells from polygon rasterization: {n_expected}"
))
# compare to db count
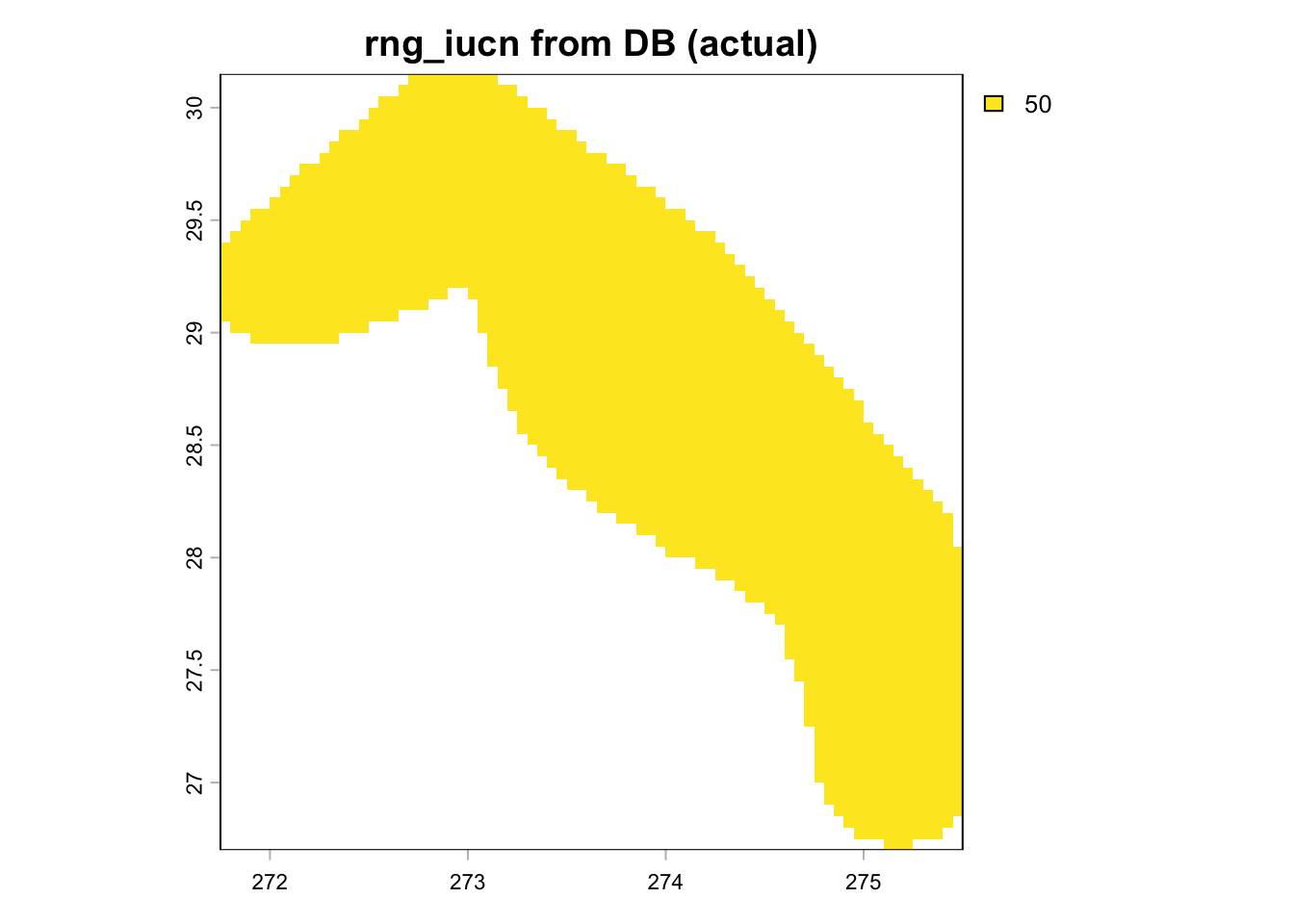
if ("rng_iucn" %in% names(r_layers)) {
n_db <- sum(!is.na(values(r_layers[["rng_iucn"]])))
message(glue(
"DB rng_iucn cells: {n_db} vs polygon rasterization: {n_expected}"
))
message(glue(
"Ratio: {round(n_db / n_expected * 100, 1)}%",
" — {ifelse(abs(n_db - n_expected) < 100, 'OK', 'CORRUPTION DETECTED')}"
))
}
# plot overlay
if (n_expected > 0) {
plot(trim(r_iucn_poly), main = "IUCN polygon rasterized (expected)")
} else {
message("No cells from polygon rasterization — CRS/extent issue")
}
if ("rng_iucn" %in% names(r_layers)) {
plot(trim(r_layers[["rng_iucn"]]), main = "rng_iucn from DB (actual)")
}
}
```
## Check for Broader Corruption
Query ALL species with rng_iucn data and check for suspicious patterns.
```{r}
#| label: broader_corruption
# use current v3 database
con_check <- cons[["v3 current"]]
# check for species with identical cell sets across datasets
d_dup_cells <- dbGetQuery(
con_check,
"
WITH ds_cells AS (
SELECT tm.taxon_id, m.ds_key, m.mdl_seq, COUNT(mc.cell_id) as n_cells
FROM taxon_model tm
JOIN model m ON tm.mdl_seq = m.mdl_seq
JOIN model_cell mc ON m.mdl_seq = mc.mdl_seq
WHERE m.ds_key IN ('rng_iucn', 'ca_nmfs', 'ch_nmfs', 'ch_fws', 'rng_fws')
GROUP BY tm.taxon_id, m.ds_key, m.mdl_seq
)
SELECT a.taxon_id, a.ds_key as ds_a, b.ds_key as ds_b,
a.n_cells, a.mdl_seq as mdl_a, b.mdl_seq as mdl_b
FROM ds_cells a
JOIN ds_cells b ON a.taxon_id = b.taxon_id
AND a.ds_key < b.ds_key
AND a.n_cells = b.n_cells
ORDER BY a.taxon_id, a.ds_key"
)
if (nrow(d_dup_cells) > 0) {
message(glue(
"{nrow(d_dup_cells)} taxon-dataset pairs with identical cell counts"
))
d_dup_cells |>
knitr::kable(
caption = "Species with identical cell counts across datasets (potential corruption)"
)
} else {
message("No species found with identical cell counts across datasets")
}
```
### Check rng_iucn cell counts for all species
```{r}
#| label: rng_iucn_counts
d_rng <- dbGetQuery(
con_check,
"
SELECT
tm.taxon_id,
t.scientific_name,
t.common_name,
t.sp_cat,
m.mdl_seq,
COUNT(mc.cell_id) as n_cells,
MIN(mc.value) as v_min,
MAX(mc.value) as v_max
FROM taxon_model tm
JOIN model m ON tm.mdl_seq = m.mdl_seq
JOIN model_cell mc ON m.mdl_seq = mc.mdl_seq
LEFT JOIN taxon t ON tm.taxon_id = t.taxon_id
WHERE m.ds_key = 'rng_iucn'
GROUP BY tm.taxon_id, t.scientific_name, t.common_name, t.sp_cat, m.mdl_seq
ORDER BY n_cells ASC"
)
message(glue(
"Total species with rng_iucn: {nrow(d_rng)}, ",
"cell count range: {min(d_rng$n_cells)} - {max(d_rng$n_cells)}"
))
# show species with suspiciously few cells (bottom 20)
d_rng |>
head(20) |>
knitr::kable(
caption = "Species with fewest rng_iucn cells (potential corruption)"
)
```
## Timeline Reconstruction
```{r}
#| label: timeline
# compare Rice's whale rng_iucn across all DB versions
d_timeline <- map_dfr(names(cons), \(db_name) {
con <- cons[[db_name]]
tryCatch(
{
d <- dbGetQuery(
con,
glue(
"
SELECT m.ds_key, COUNT(mc.cell_id) as n_cells,
MIN(mc.value) as v_min, MAX(mc.value) as v_max
FROM taxon_model tm
JOIN model m ON tm.mdl_seq = m.mdl_seq
JOIN model_cell mc ON m.mdl_seq = mc.mdl_seq
WHERE tm.taxon_id = {taxon_id_ricei}
AND m.ds_key IN ('rng_iucn', 'ca_nmfs', 'ch_nmfs', 'ms_merge')
GROUP BY m.ds_key"
)
)
d$db_version <- db_name
d
},
error = \(e) tibble()
)
})
if (nrow(d_timeline) > 0) {
d_timeline |>
select(db_version, ds_key, n_cells, v_min, v_max) |>
arrange(db_version, ds_key) |>
knitr::kable(
caption = "Rice's whale cell counts across DB versions (timeline)"
)
} else {
message("No timeline data found")
}
```
```{r}
#| label: timeline_notes
message(
"
Timeline of key events:
- Jan 22-23: IUCN range rasterization (6.2 hrs)
- Jan 29: backup sdm_2026-01-29.duckdb
- Feb 10: backup sdm_2026-02-10.duckdb
- Feb 11: fix_taxon_model_dups.qmd — cleaned duplicates
- Feb 12: calc_scores.qmd recalculated — backup sdm_v3_2026-02-12.duckdb
If Jan 29 backup already shows ~1,969 cells for rng_iucn, then the corruption
happened during or immediately after IUCN range rasterization (Jan 22-23).
Check if the rasterized TIF was correct (sf::summarize() with sf_use_s2(FALSE)
may have produced GEOMETRYCOLLECTION instead of valid polygon).
"
)
```
## Cleanup
```{r}
#| label: disconnect
walk(cons, \(con) dbDisconnect(con, shutdown = TRUE))
```