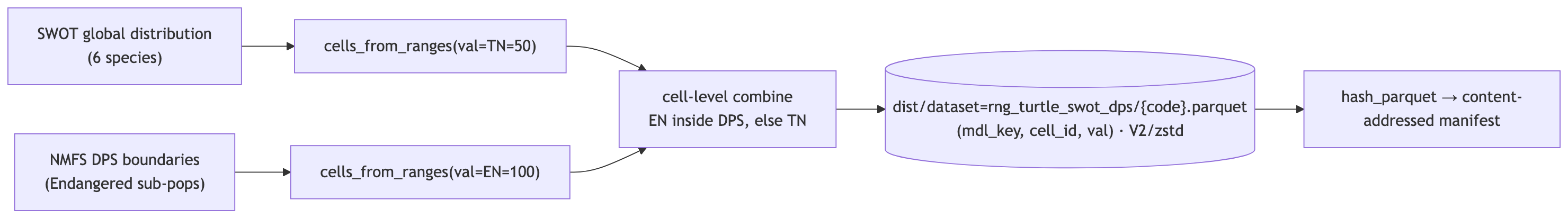
Ingest SWOT + NMFS-DPS sea turtle ranges → global 0.05° cells
Refined sea-turtle ranges onto the global 0.05° cell grid, replacing the overly broad AquaMaps/IUCN turtle ranges. Range polygons come from SWOT global distributions; NMFS DPS boundaries mark the Endangered sub-populations.
Value = extinction-risk score via msens::compute_er_score() (never hard-coded). For the three species with DPS data (Loggerhead CC, Green CM, Olive Ridley LO): cells inside an Endangered DPS → compute_er_score("NMFS:EN") = 100, the rest of the range (Threatened) → compute_er_score("NMFS:TN") = 50. The three pure-Endangered species (Leatherback DC, Hawksbill EI, Kemp’s Ridley LK): 100 throughout. The EN/TN split is done at the cell level (rasterize both, combine) — no fragile geometry intersection. Whole range, no land mask (turtles are marine, so these are ocean cells). mdl_key = "rng_turtle_swot_dps|{code}".
1 Design
2 Setup
Code
librarian::shelf(
arrow, DBI, dplyr, duckdb, fs, glue, here, jsonlite, logger, readr, sf, terra,
tibble, MarineSensitivity/msens, quiet = T)
source(here("libs/paths.R")) # dir_raw, dir_big_v, cellid_tif, ver
source(here("libs/vars.R")) # redo_ingest
options(readr.show_col_types = F)
ds_key <- "rng_turtle_swot_dps"
dir_swot <- glue("{dir_raw}/swot_seamap.env.duke.edu/swot_distribution")
dir_dps <- glue("{dir_raw}/fisheries.noaa.gov/turtle-DPS_endangered")
dir_dist <- glue("{dir_big_v}/marine-atlas/dist/dataset={ds_key}")
manifest <- here("data/manifests/ingest_turtles_swot_dps.json")
dir_create(c(dir_dist, path_dir(manifest)))
stopifnot("run build_cell_grid.qmd first" = file_exists(cellid_tif), dir_exists(dir_swot))
# range value = extinction-risk score via msens::compute_er_score() (NEVER hard-code).
# Sea turtles are ESA-listed marine species (NMFS authority): EN=100, TN=50.
er_en <- compute_er_score("NMFS:EN") # 100
er_tn <- compute_er_score("NMFS:TN") # 50
swot_spp <- tribble(
~code, ~sci_name, ~common_name, ~has_dps, ~esa_pure,
"CC", "Caretta caretta", "Loggerhead", TRUE, NA,
"CM", "Chelonia mydas", "Green", TRUE, NA,
"DC", "Dermochelys coriacea", "Leatherback", FALSE, "EN",
"EI", "Eretmochelys imbricata", "Hawksbill", FALSE, "EN",
"LK", "Lepidochelys kempii", "Kemp's Ridley", FALSE, "EN",
"LO", "Lepidochelys olivacea", "Olive Ridley", TRUE, NA) |>
mutate(mdl_key = mdl_key_raw(ds_key, code))
write_csv(swot_spp, glue("{dir_dist}/../model_{ds_key}.csv"))3 Rasterize each range (EN=100 / TN=50) → one Parquet each
Code
if (redo_ingest && dir_exists(dir_dist)) { dir_delete(dir_dist); dir_create(dir_dist) }
for (i in seq_len(nrow(swot_spp))) {
s <- swot_spp[i, ]
out_pq <- fs::path(dir_dist, s$code, ext = "parquet")
if (file_exists(out_pq)) { log_info("{s$code}: skip (exists)"); next }
swot <- st_read(glue("{dir_swot}/Global_Distribution_{s$code}.shp"), quiet = TRUE)
if (s$has_dps) {
dps <- st_read(glue("{dir_dps}/{tolower(s$code)}_endangered.gpkg"), quiet = TRUE)
d_range <- cells_from_ranges(swot, cellid_tif, value = er_tn) # whole range = Threatened
d_en <- cells_from_ranges(dps, cellid_tif, value = er_en) # Endangered DPS areas
d <- d_range |> mutate(val = if_else(cell_id %in% d_en$cell_id, er_en, er_tn))
} else {
d <- cells_from_ranges(swot, cellid_tif, value = er_en) # pure Endangered
}
if (nrow(d) == 0) { log_warn("{s$code}: no cells"); next }
msens::write_atlas_parquet(tibble(mdl_key = s$mdl_key, cell_id = d$cell_id, val = d$val), out_pq)
log_info("{s$code} ({s$common_name}): {nrow(d)} cells")
}Spherical geometry (s2) switched offSpherical geometry (s2) switched onSpherical geometry (s2) switched offSpherical geometry (s2) switched onSpherical geometry (s2) switched offSpherical geometry (s2) switched onSpherical geometry (s2) switched offSpherical geometry (s2) switched onSpherical geometry (s2) switched offSpherical geometry (s2) switched onSpherical geometry (s2) switched offSpherical geometry (s2) switched onSpherical geometry (s2) switched offSpherical geometry (s2) switched onSpherical geometry (s2) switched offSpherical geometry (s2) switched onSpherical geometry (s2) switched offSpherical geometry (s2) switched on4 Verify
Code
pq <- dir_ls(dir_dist, glob = "*.parquet")
con <- dbConnect(duckdb())
smry <- dbGetQuery(con, glue(
"SELECT replace(mdl_key,'rng_turtle_swot_dps|','') code, count(*) n_cells,
count(*) FILTER (WHERE val = 100) n_en, count(*) FILTER (WHERE val = 50) n_tn
FROM read_parquet('{dir_dist}/*.parquet') GROUP BY mdl_key ORDER BY code"))
# order-independent content fingerprint of the on-disk surface (not the ingest run)
h <- msens::hash_parquet(glue("{dir_dist}/*.parquet"), con)
dbDisconnect(con, shutdown = TRUE)
msens::report_table(smry, caption = "rng_turtle_swot_dps model_cell surface")| code | n_cells | n_en | n_tn |
|---|---|---|---|
| CC | 20339916 | 10945504 | 9394412 |
| CM | 18244111 | 4461588 | 13782523 |
| DC | 24548372 | 24548372 | 0 |
| EI | 13410951 | 13410951 | 0 |
| LK | 1110528 | 1110528 | 0 |
| LO | 14954346 | 5845514 | 9108832 |
5 Manifest (target output)
Code
# content-addressed manifest: deterministic, no wall-clock, no machine paths ->
# downstream targets re-run only when this surface's content actually changes
msens::write_manifest(
manifest, target = "ingest_turtles_swot_dps", content_hash = h,
stats = list(ds_key = ds_key, ver = ver, n_species = nrow(swot_spp),
n_parquet = length(pq)),
force = msens::force_target("ingest_turtles_swot_dps"))