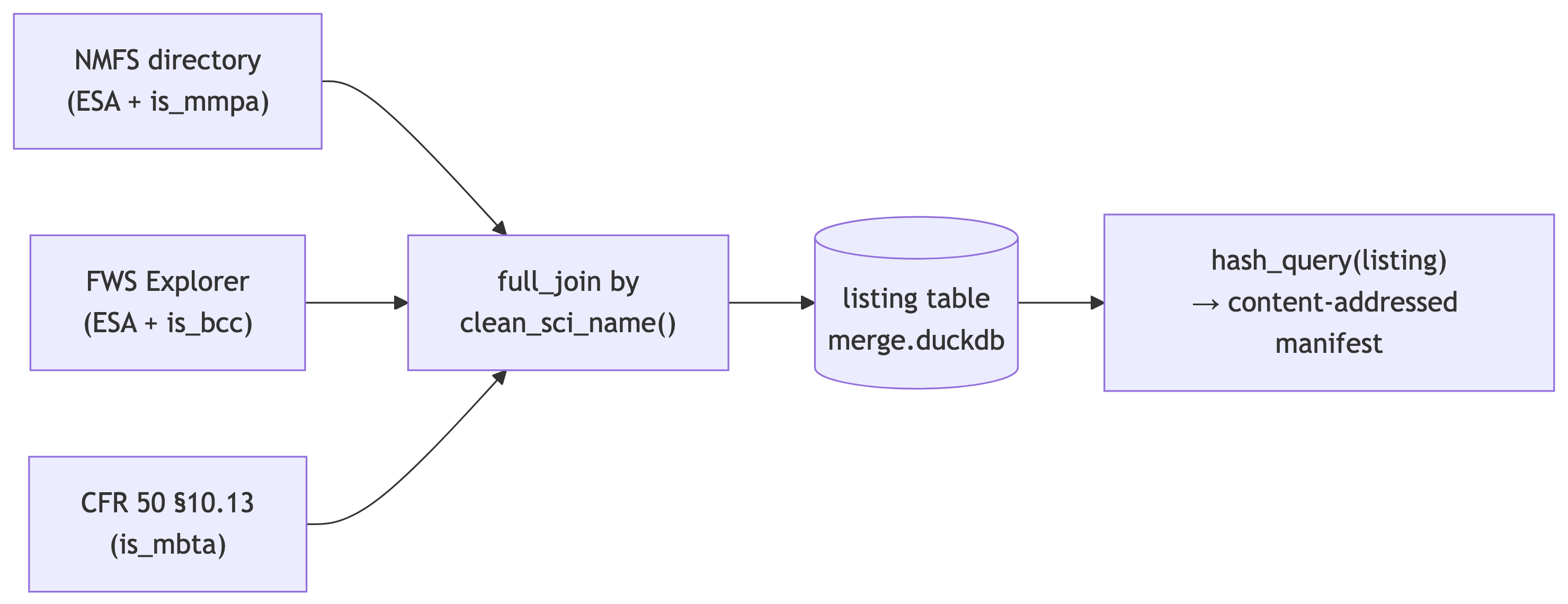
Ingest US federal listings → MMPA / MBTA / ESA taxon attributes
Read the US federal species lists and derive per-species attributes that drive the taxon-level extinction-risk score (compute_er_score floors): ESA status (NMFS + FWS), is_mmpa (Marine Mammal Protection Act, from the NMFS directory), is_mbta (Migratory Bird Treaty Act, from the FWS 10.13 list — essential now that v8 ingests all birds, not v7’s 571), and is_bcc (Birds of Conservation Concern). Keyed by clean_sci_name() so the merge crosswalk joins it to each taxon; the US-vs-IUCN precedence + compute_er_score happen in merge_taxon.
1 Design
2 Setup + read the three lists
Code
librarian::shelf(DBI, dplyr, duckdb, fs, glue, here, janitor, jsonlite, logger,
readr, readxl, stringr, tibble, MarineSensitivity/msens, quiet = T)
source(here("libs/paths.R"))
options(readr.show_col_types = F)
nmfs_csv <- here("data/nmfs_species-directory.csv")
fws_csv <- glue("{dir_raw}/fws.gov/species/FWS_Species_Data_Explorer.csv")
mbta_xlsx <- glue("{dir_raw}/fws.gov/birds_mbta/CFR50-Part10.13-2023.xlsx")
merge_db <- glue("{dir_big_v}/marine-atlas/merge.duckdb")
manifest <- here("data/manifests/ingest_listings.json")
dir_create(path_dir(manifest))
stopifnot(all(file_exists(c(nmfs_csv, fws_csv, mbta_xlsx))))
# NMFS species directory -> ESA status (NMFS) + is_mmpa (parse_noaa_status)
d_nmfs_raw <- read_csv(nmfs_csv)
d_nmfs <- d_nmfs_raw |>
bind_cols(parse_noaa_status(d_nmfs_raw$protected_status)) |>
transmute(sci = clean_sci_name(scientific_name), nmfs_esa = esa_status, is_mmpa) |>
group_by(sci) |>
summarise(nmfs_esa = if (any(nmfs_esa == "EN")) "EN" else if (any(nmfs_esa == "TN")) "TN" else "LC",
is_mmpa = any(is_mmpa), .groups = "drop")
# FWS Data Explorer -> ESA status (FWS) + is_bcc
d_fws <- read_csv(fws_csv) |> clean_names() |>
transmute(
sci = clean_sci_name(scientific_name),
fws_esa = case_when(str_detect(esa_listing_status, "Endangered") ~ "EN",
str_detect(esa_listing_status, "Threatened") ~ "TN", TRUE ~ "LC"),
is_bcc = is_bcc %in% c(TRUE, "TRUE")) |>
group_by(sci) |>
summarise(fws_esa = if (any(fws_esa == "EN")) "EN" else if (any(fws_esa == "TN")) "TN" else "LC",
is_bcc = any(is_bcc), .groups = "drop")
# MBTA 10.13 list -> is_mbta (Genus + species binomial in cols 4-5)
mbta <- read_excel(mbta_xlsx, sheet = "10.13 List", skip = 3, col_names = FALSE)New names:
• `` -> `...1`
• `` -> `...2`
• `` -> `...3`
• `` -> `...4`
• `` -> `...5`
• `` -> `...6`
• `` -> `...7`
• `` -> `...8`Code
mbta_sci <- clean_sci_name(paste(mbta[[4]], mbta[[5]])) |> unique()3 Combine → listing table (keyed by clean scientific name)
Code
listing <- full_join(d_nmfs, d_fws, by = "sci") |>
mutate(
is_mmpa = coalesce(is_mmpa, FALSE),
is_bcc = coalesce(is_bcc, FALSE),
is_mbta = sci %in% mbta_sci,
nmfs_esa = coalesce(nmfs_esa, "LC"),
fws_esa = coalesce(fws_esa, "LC")) |>
# add MBTA-only birds not otherwise in the NMFS/FWS tables
bind_rows(tibble(sci = setdiff(mbta_sci, full_join(d_nmfs, d_fws, by = "sci")$sci),
nmfs_esa = "LC", fws_esa = "LC",
is_mmpa = FALSE, is_bcc = FALSE, is_mbta = TRUE)) |>
distinct(sci, .keep_all = TRUE)
con <- dbConnect(duckdb(merge_db))
dbWriteTable(con, "listing", listing, overwrite = TRUE)
smry <- dbGetQuery(con, "SELECT count(*) n, sum(is_mmpa::int) mmpa, sum(is_mbta::int) mbta,
sum(is_bcc::int) bcc, count(*) FILTER(WHERE nmfs_esa<>'LC' OR fws_esa<>'LC') esa_listed FROM listing")
# content fingerprint of the listing table (before disconnect)
h <- msens::hash_query(con, "listing")
dbDisconnect(con, shutdown = TRUE)
msens::report_table(smry, caption = "US federal listing table summary")| n | mmpa | mbta | bcc | esa_listed |
|---|---|---|---|---|
| 11532 | 70 | 1104 | 320 | 2402 |
4 Manifest
Code
# content-addressed manifest: deterministic (no wall-clock) -> downstream targets
# re-run only when the listing table's content actually changes
msens::write_manifest(
manifest, target = "ingest_listings", content_hash = h,
stats = list(ver = ver, n = smry$n, n_mmpa = smry$mmpa,
n_mbta = smry$mbta, n_bcc = smry$bcc),
force = msens::force_target("ingest_listings"))